
Abstract
The hippocampus is an important brain region that is involved in neurological disorders such as Alzheimer disease, schizophrenia, and epilepsy. Ionotropic glutamate receptors—namely, N-methyl-D-aspartate (NMDA) receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors (AMPARs), and kainic acid (KA) receptors (KARs)—are well known to be involved in these diseases by mediating long-term potentiation, excitotoxicity, or both. To predict the therapeutic efficacy and neuronal toxicity of drug candidates acting on these receptors, physiologically relevant systems for assaying brain region–specific human neural cells are necessary. Here, we characterized the functional differentiation of human fetal hippocampus–derived neural stem/progenitor cells—namely, HIP-009 cells. Calcium rise assay demonstrated that, after a 4-week differentiation, the cells responded to NMDA (EC50 = 7.5 ± 0.4 µM; n = 4), AMPA (EC50 = 2.5 ± 0.1 µM; n = 3), or KA (EC50 = 33.5 ± 1.1 µM; n = 3) in a concentration-dependent manner. An AMPA-evoked calcium rise was observed in the absence of the desensitization inhibitor cyclothiazide. In addition, the calcium rise induced by these agonists was inhibited by antagonists for each receptor—namely, MK-801 for NMDA stimulation (IC50 = 0.6 ± 0.1 µM; n = 4) and NBQX for AMPA and KA stimulation (IC50 = 0.7 ± 0.1 and 0.7 ± 0.03 µM, respectively; n = 3). The gene expression profile of differentiated HIP-009 cells was distinct from that of undifferentiated cells and closely resembled that of the human adult hippocampus. Our results show that HIP-009 cells are a unique tool for obtaining human hippocampal neural cells and are applicable to systems for assay of ionotropic glutamate receptors as a physiologically relevant in vitro model.
Keywords
Introduction
Hippocampal neurons play critical roles in learning and memory functions in the central nervous system (CNS). Long-term potentiation is thought to be the mechanism behind memory formation and is mediated through the activation of glutamate receptors. Glutamate receptors are also responsible for excitotoxicity-mediated neuronal death. Excitotoxicity underlies the pathology of a number of neurological abnormalities associated with Huntington disease (HD), Alzheimer disease (AD), and Parkinson disease (PD).1,2 Glutamate receptors are classified into two general groups: metabotropic and ionotropic. Metabotropic receptors are involved in the expression of slow postsynaptic potentials through guanosine triphosphate (GTP)– binding proteins as second messengers. 3 Ionotropic receptors mediate fast postsynaptic potentials directly through activated ion channels. 4 Within the ionotropic family of glutamate receptors, there are three subtypes based on the type of synthetic agonist for each receptor—namely, N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), and kainic acid (KA) receptors. Under physiological conditions, each receptor is formed by complex and diverse assemblies of subunits. 5 The NMDA receptor (NMDAR) family is composed of seven subunits: GluN1, GluN2A−D, GluN3A, and GluN3B. Most NMDARs contain two GluN1 subunits plus two GluN2 subunits. The AMPA receptors (AMPARs) are tetrameric aggregates of one or more of four subunits (GluA1−4). In addition, neuronal AMPARs contain transmembrane AMPA receptor regulatory proteins (TARPs), which modulate desensitization. 6 The KA receptor (KAR) family consists of five genes encoding five subunits (GluK1−5). Similar to NMDARs and AMPARs, KARs are tetramers.
Despite the fact that under physiological conditions, each ionotropic glutamate receptor has complex combinations of subunits and associated proteins, such as the TARPs included in AMPARs, gene-overexpressing cells are generally used for functional in vitro screening in drug discovery programs. Because such cells usually overexpress the genes encoding one or a few target proteins, this drug discovery approach can focus on only the target protein of interest. In parallel with this approach, an assay system that mimics the complexity of physiological conditions is necessary to predict the clinical efficacy and adverse effect of drug candidates. As a solution, primary neural cells of rodents have been used in many in vitro assays; because of their naturally occurring protein expression levels and subunit combinations, such cells are more physiologically relevant than cells artificially overexpressing genes. There is, however, concern about species differences between rodents and humans. Therefore, human neural cells are anticipated to be a novel and ideal tool for investigating the clinical effects of drug candidates in vitro.7,8
Generation of human embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) and the development of neural differentiation methods by using these cells have expanded opportunities to generate human neural cells. However, there are lots of reported methods for differentiating human ESCs and iPSCs into functional neurons, and the differentiation methods differ from laboratory to laboratory. 9 Accordingly, there has not been a standardized method yet by which everyone could obtain reproducible data easily. These issues are critical hurdles if these methods are to be applied to drug screening. In addition, methods of brain region–specific neural differentiation from human ESCs/iPSCs have not been fully established, and methods for directed differentiation of hippocampal neurons from human ESCs/iPSCs are under development. The hippocampus is an important brain region involved in neurological disorders such as AD, schizophrenia, and epilepsy.10–13 Therefore, we need to develop an assay system that uses human hippocampal neurons so that we can obtain a detailed understanding of the molecular mechanisms underlying these diseases and discover novel drugs for treating them.
There is an additional promising tool for using human hippocampal neural cells—namely, human hippocampal neural stem/progenitor cells. Here, we examined the ability of HIP-009 cells, which are human fetal hippocampus–derived neural stem/progenitor cells, to differentiate into hippocampal neural cells. We also demonstrated the functional expression of NMDARs, AMPARs, and KARs in differentiated HIP-009 cells. We found that human hippocampal neural cells derived from HIP-009 cells are a novel tool for use in the in vitro brain region–specific and physiologically relevant systems for assaying human ionotropic glutamate receptors.
Materials and Methods
Chemicals
NMDA, (+)-MK-801 hydrogen maleate, memantine hydrochloride, D-serine, cyclothiazide (CTZ), and NBQX hydrate were purchased from Sigma-Aldrich (St. Louis, MO). D-(–)- 2-Amino-5-phosphonopentanoic acid (D-AP5), 7-chlorokynurenic acid (7-Cl-KYNA), AMPA, KA, and CNQX disodium salt were obtained from Tocris Bioscience (Bristol, UK). Sodium hydrogen L(+)-glutamate monohydrate, carbamazepine, and glycine were purchased from Wako Pure Chemical Industries (Osaka, Japan).
Cell Culture
HIP-009 cells were purchased from GigaCyte, LLC (Branford, CT; now PhoenixSongs Biologicals, Branford, CT). Cells were expanded and differentiated as described in the manufacturer’s instructions. Briefly, cells were proliferated on laminin (Sigma-Aldrich)–coated dishes in Neural StemCell Growth Medium (PhoenixSongs Biologicals). Proliferated cells were split for expansion every 4 or 5 days. Before the start of differentiation, expanded cells were plated on laminin-coated plates in Neural Transition Medium (PhoenixSongs Biologicals) and cultured for 3 days. After that, cells were seeded on poly-D-lysine (PDL; Sigma-Aldrich)–coated plates in Neural Differentiation Medium (PhoenixSongs Biologicals) for differentiation. The differentiation process was performed for 4 weeks, during which half of the medium was changed twice a week. All cell culture was performed at 37 °C in a humidified atmosphere of 2% O2 and 6% CO2. HIP-009 cells at passages 8 to 15 were used. This study was approved by the Eisai Research Ethics Committee.
Immunocytochemistry
Cells were washed with phosphate-buffered saline (PBS) without calcium and magnesium (Wako Pure Chemical Industries) once, then fixed in 1% to 2% paraformaldehyde (Wako Pure Chemical Industries) for 30 min at room temperature. The cells were then washed with PBS at room temperature three times and permeabilized with 0.2% Triton X-100/PBS for 5 min at room temperature. Nonspecific binding was blocked with Blocking One (Nacalai Tesque, Kyoto, Japan) in PBS (blocking buffer) for 60 min at room temperature. Cells were incubated overnight at 4 °C with primary antibodies diluted in 0.2% Triton X-100/blocking buffer. After being washed with 0.05% Tween 20/PBS (PBST) for 5 min three times, cells were incubated for 1 h at room temperature with secondary antibodies and Hoechst 33342 (1 µg/mL; Sigma-Aldrich) diluted in 0.2% Triton X-100/blocking buffer. Cells were then washed with PBST three times and rinsed with PBS.
The primary antibodies used were as follows: mouse–anti-nestin (1:1000; Abcam, Cambridge, UK), goat–anti-SOX1 (1:500; R&D Systems, Minneapolis, MN), mouse–anti-PAX6 (1:100; Abcam), mouse–anti-Ki67 (1:100; BD Pharmingen, San Diego, CA), mouse–anti-βIII-tubulin (1:1000; Covance Research Products, Denver, PA), mouse–anti-MAP2 (1:1000; Sigma-Aldrich), mouse–anti-Hu C/D (1:100; Life Technologies, Carlsbad, CA), rabbit–anti-GFAP (1:1000; DAKO, Glostrup, Denmark), mouse–anti-CNPase (1:500; Sigma-Aldrich), rabbit–anti-GABA (1:1000; Sigma-Aldrich), and guinea pig–anti-vGlut1 (1:1000; Millipore, Billerica, MA).
The secondary antibodies were Alexa Fluor 488 donkey anti–mouse IgG, Alexa Fluor 488 donkey anti–goat IgG, Alexa Fluor 488 donkey anti–rabbit IgG, Alexa Fluor 594 donkey anti–rabbit IgG (Life Technologies), DyLight 594 donkey anti–guinea pig IgG, and DyLight 649 donkey anti–mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). All secondary antibodies were used at 1:500 dilutions. Cells were imaged under a laser-scanning confocal microscope (CellVoyager; Yokogawa Electric Corp., Tokyo, Japan). Images were taken by using a maximum intensity projection method. For population analysis, the number of Hu C/D- or GFAP-positive cells as a percentage of the total number of cells was calculated.
Quantitative Reverse Transcription PCR
For quantitative reverse transcription PCR (RT-qPCR) analysis, RNAs were isolated with an RNeasy Mini Kit (Qiagen, Venlo, Netherlands), treated with DNase (Qiagen), and reverse-transcribed into complementary DNAs (cDNAs) with SuperScript VILO Master Mix (Life Technologies) in accordance with the manufacturer’s instructions. RT-qPCR experiments were performed by using an ABI 7900HT System (Life Technologies) with EagleTaq Master Mix with ROX (Roche Diagnostics, Indianapolis, IN) and TaqMan primers and probe sets (Life Technologies). The thermocycling protocol consisted of an initial cycle at 95 °C for 10 min, followed by 40 cycles of PCR at 95 °C for 15 s and 60 °C for 1 min. The messenger RNA (mRNA) level of each sample was normalized against the mRNA expression of 18S from the same sample, and data were expressed as fold change compared with undifferentiated HIP-009 cells.
Microarray Analysis
In total, 200 ng of total RNA from each cell culture and human hippocampus (Clontech Laboratories, Inc., Mountain View, CA) was reverse-transcribed into double-stranded cDNAs and amplified for 2 h at 40 °C by using a Low Input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA). The cDNAs were then transcribed into antisense complementary RNAs (cRNAs) and labeled with Cy3-CTP fluorescent dyes for 2 h at 40 °C in accordance with the manufacturer’s protocol. Cy3-labeled cRNAs were purified with an RNeasy Mini Kit (Qiagen). Purity and dye incorporation were assessed with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and 2100 Bioanalyzer (Agilent Technologies). The minimum acceptable Cy3 labeling was ≥6.0 pmol/µg. Next, 600 ng of Cy3-RNAs was hybridized to a SurePrint G3 Human GE Microarray kit 8 × 60K ver. 1.0 by using a Gene Expression Hybridization Kit (Agilent Technologies). After incubation for 17 h at 65 °C and 10 rpm rotation, the microarrays were washed with Gene Expression Wash Buffer 1 (Agilent Technologies) for 1 min at room temperature and then with Wash Buffer 2 (Agilent Technologies) for 1 min at 37 °C. The microarrays were scanned with a G2505B Microarray Scanner (Agilent Technologies). Data were analyzed with GeneSpring 12.5 software (Agilent Technologies).
Electrophysiology
For electrophysiological recordings, HIP-009 cells were seeded on glass coverslips coated with laminin and PDL. After 4 weeks of differentiation, the cells were characterized by using whole-cell patch-clamp recording techniques. Signals were low pass filtered at 3 kHz and sampled at 20 kHz by using a Digidata 1322A interface (Molecular Devices, Sunnyvale, CA). Data recording and analysis were performed with pCLAMP 10.2 software (Molecular Devices). The extracellular solution contained 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 24 mM D-glucose (pH adjusted to 7.4 with NaOH). The intracellular solution contained 130 mM KCl, 1 mM EGTA, 1 mM MgCl2, 5 mM HEPES, and 5 mM Na2-ATP (pH adjusted to 7.2 with KOH). Patch micropipettes were made from borosilicate glass capillaries (Drummond Scientific, Broomall, PA) by using a PB-7 pipette puller (Narishige, Tokyo, Japan). Current-clamp and voltage-clamp recordings were performed with an Axopatch 200B amplifier (Molecular Devices). For the current-clamp recordings, current pulses were injected through the patch electrode into differentiated HIP-009 cells with a 10-pA step increment from −10 pA to +100 pA. For the voltage-clamp recordings, cells were subjected to 20-mV step depolarizations from −80 mV to +80 mV at a −100-mV holding potential.
Fluorescence-Based Calcium Rise Assay
For the assays of differentiated HIP-009 cells, HIP-009 cells cultured in Neural Transition Medium for 3 days were plated on black-walled, clear-bottomed, PDL-coated 96-well plates (Corning Incorporated, Corning, NY) at 3.7 × 104 cells/well in Neural Differentiation Medium and then allowed to differentiate into neural cells for 4 weeks. Undifferentiated HIP-009 cells were seeded on the same types of 96-well plates (which also had been coated manually with laminin) at 6.4 × 103 cells/well in StemCell Growth Medium and were cultured until they reached 80% to 100% confluence. On the day of the assays, the media were removed and the cells were loaded with calcium 4 dye (for glutamate concentration–dependent assays) or calcium 5 dye (for the other assays) (Molecular Devices) and probenecid (2.5 mM; Life Technologies) reconstituted in an assay buffer for 1 h at 37 °C. The compositions of the assay buffers were as follows: 20 mM HEPES and Hank’s balanced salt solution with calcium and magnesium without phenol red (Life Technologies) (pH adjusted to 7.4 with NaOH) for glutamate concentration–dependent assays; 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 10 mM HEPES, and 10 mM D-glucose (pH adjusted to 7.4 with NaOH) for NMDARs and co-treatment assays of MK-801 and NBQX; and 140 mM NaCl, 5 mM KCl, 3 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 24 mM D-glucose, and 10 µM MK-801 (pH adjusted to 7.4 with NaOH) for AMPARs and KARs. Compounds were diluted in the assay buffer and transferred to compound plates (Thermo Fisher Scientific). After the 1-h incubation, except in the case of the glutamate concentration–dependent assays, the cells were washed twice and replaced with the assay buffer. In the case of the glutamate concentration–dependent assays, the washing step was skipped. Calcium rise was measured with a Functional Drug Screening System 6000 (FDSS 6000; Hamamatsu Photonics, Hamamatsu, Japan) that simultaneously monitored changes in fluorescence in each well of the plate. A baseline was recorded for 12 s before the addition of the compound solution, and recordings were taken at 0.3-s intervals for 1.75 min in total (excitation wavelength, 480 nm; emission, 540 nm).
Statistical Analysis
All data were expressed as means ± standard errors of means (SEM); n refers to the number of independent experiments. GraphPad Prism 6.0 (GraphPad Software, San Diego, CA) was used for statistical analysis. Statistical differences between two groups were conducted by use of the F test, followed by the unpaired Student t test (p ≥ 0.05 in F test) or Welch’s t test (p < 0.05 in F test). In the studies of the effects of the combination of NBQX and MK-801 on inhibition of the intracellular glutamate-evoked calcium rise, two-way analysis of variance (ANOVA) was performed on the percent values of the glutamate responses. Tukey’s multiple-comparison test was used to examine the differences between two groups. A probability (p) value of < 0.05 was considered statistically significant.
Results
Immunocytochemical Characterization of HIP-009 Cells
To verify that HIP-009 cells had the characteristics of stem/progenitor cells before differentiation, undifferentiated HIP-009 cells were examined with markers for neural stem/progenitor cells and for proliferation immunocytochemically. Almost all cells were positive for the neural stem/progenitor markers nestin, PAX6, and SOX1, as well as for the proliferation marker Ki67 (
Fig. 1A
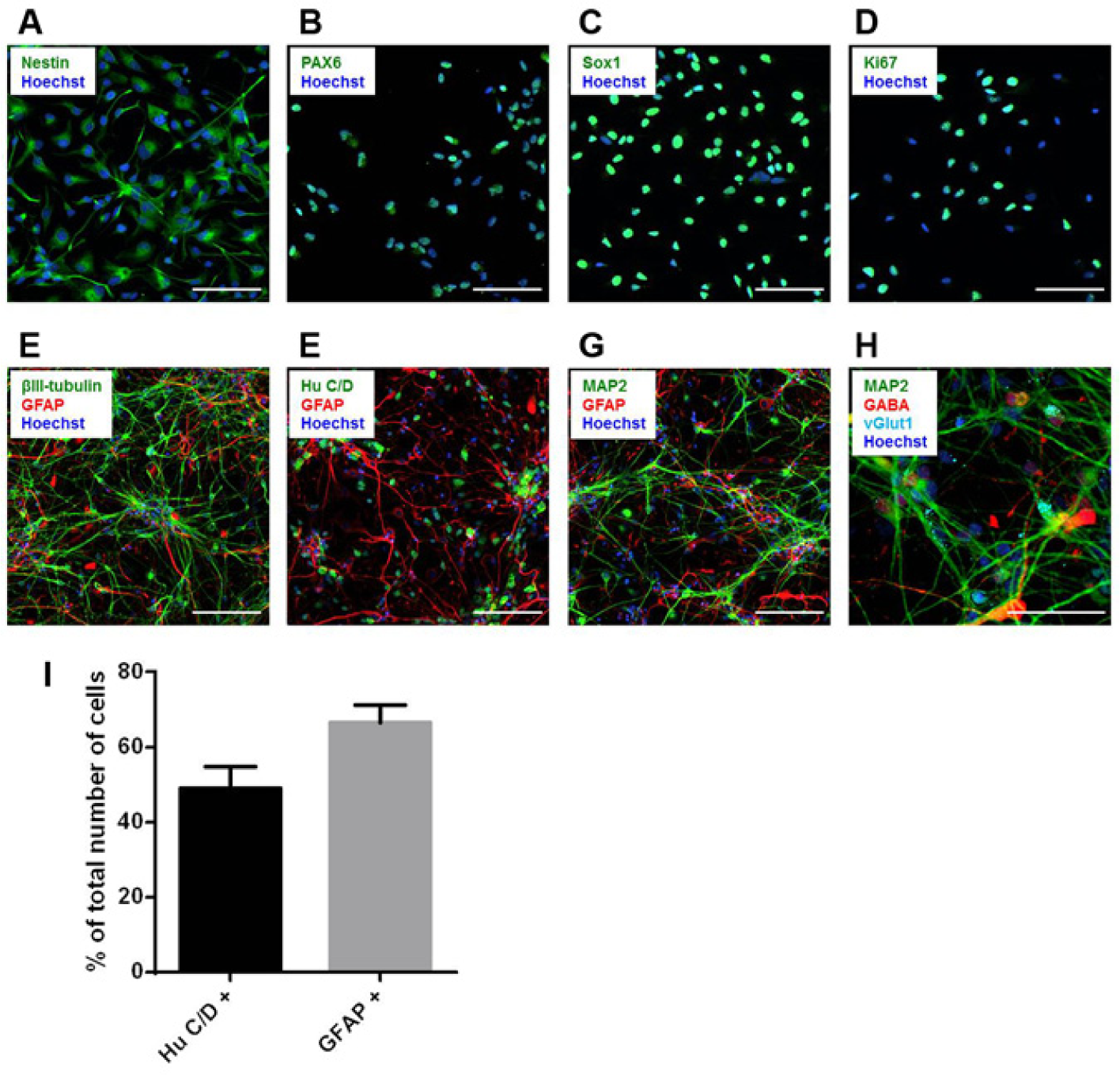
Immunocytochemical characterization of HIP-009 cells. Undifferentiated HIP-009 cells were immunostained with the neural stem/progenitor markers nestin (
Gene Expression Analysis of HIP-009 Cells
To investigate the gene expression profiles of HIP-009 cells, we conducted a microarray analysis of the undifferentiated and differentiated cells. Two-dimensional principal component analysis revealed that the gene expression profile of differentiated HIP-009 cells was distinct from that of undifferentiated cells and closely resembled that of the human adult hippocampus (
Fig. 2A
). These findings were also especially applicable to neural markers (
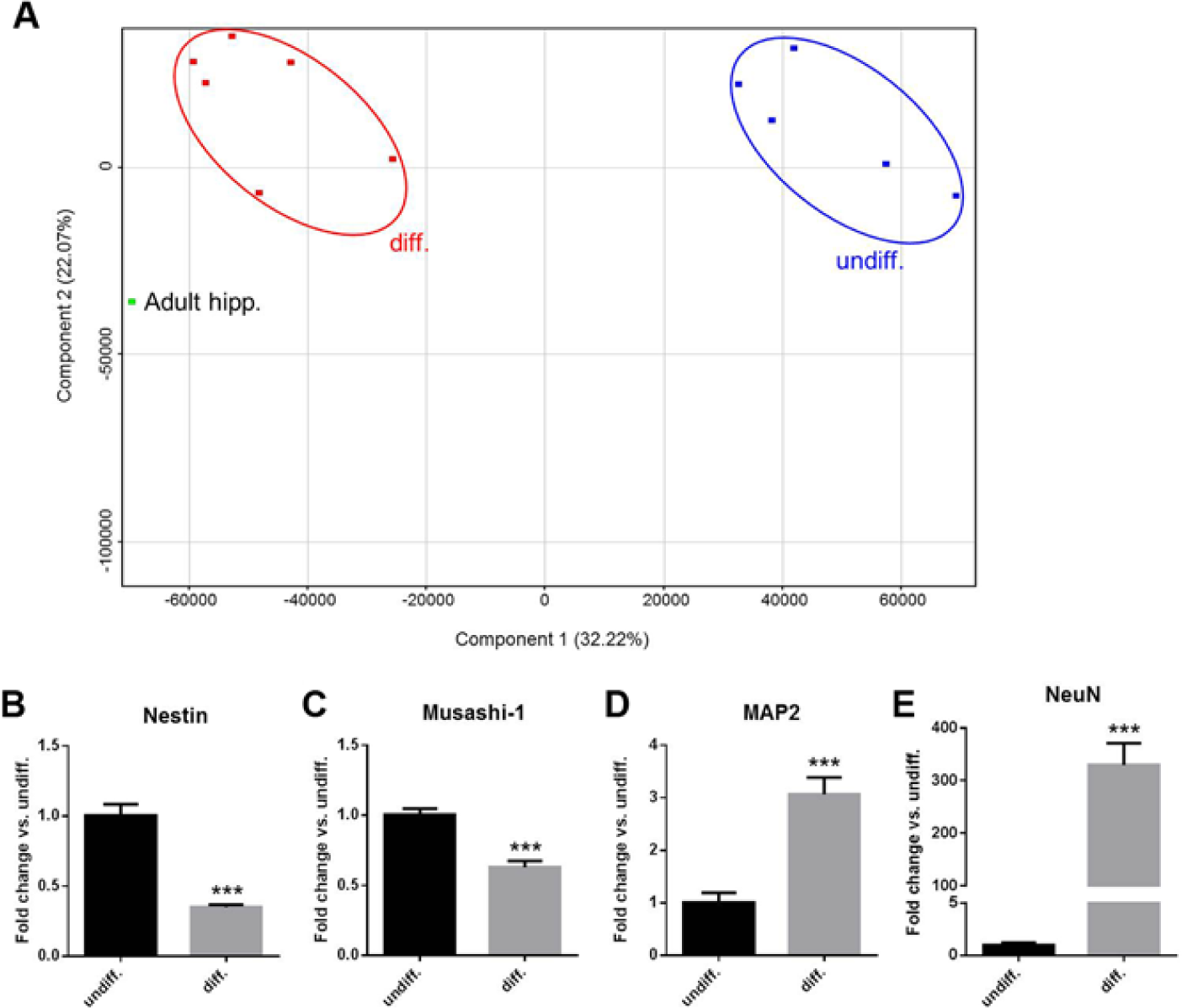
Gene expression analysis of HIP-009 cells. (
Electrophysiological Analysis of Differentiated HIP-009 Cells
Functional maturation of differentiated HIP-009 neurons was determined by patch-clamp analysis. Voltage-clamp analysis revealed that voltage-dependent currents were invoked ( Fig. 3A ), and the dynamics of the inward and outward currents were consistent with those of sodium and delayed rectifier potassium currents, respectively. Current-clamp recordings of differentiated HIP-009 neurons demonstrated that the cells fired action potentials in response to depolarizing current injections ( Fig. 3B ).
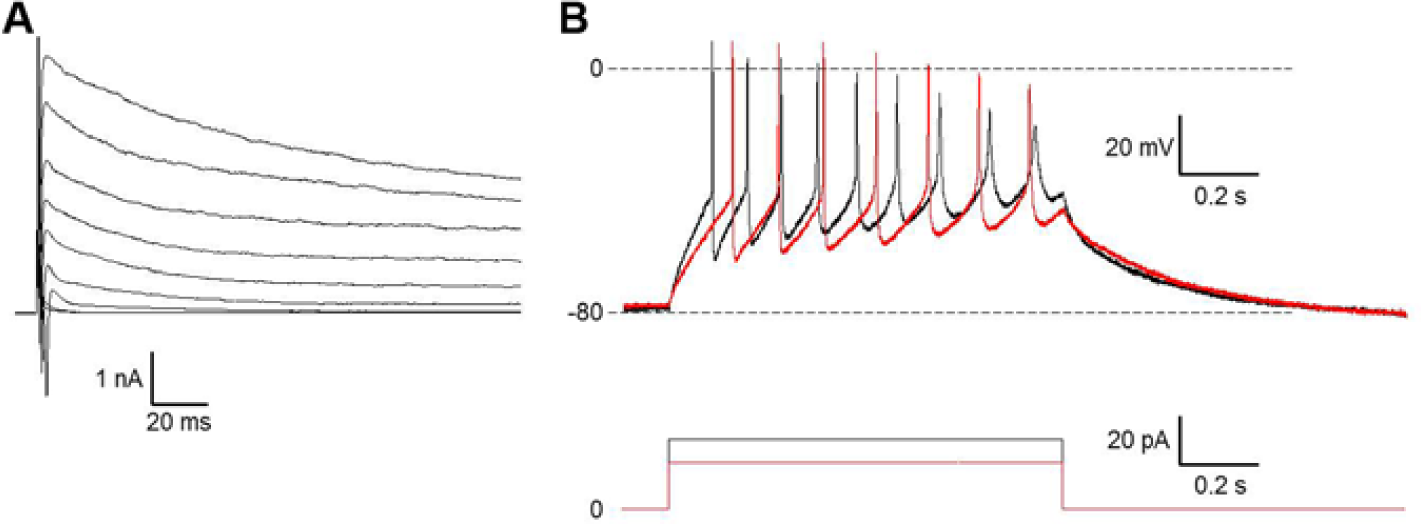
Electrophysiological recordings from differentiated HIP-009 cells. (
Identification of Gene Expression of Ionotropic Glutamate Receptors in HIP-009 Cells
In the microarray analysis, genes encoding ionotropic glutamate receptors (NMDARs, AMPARs, and KARs) were upregulated in differentiated HIP-009 cells compared with undifferentiated cells (
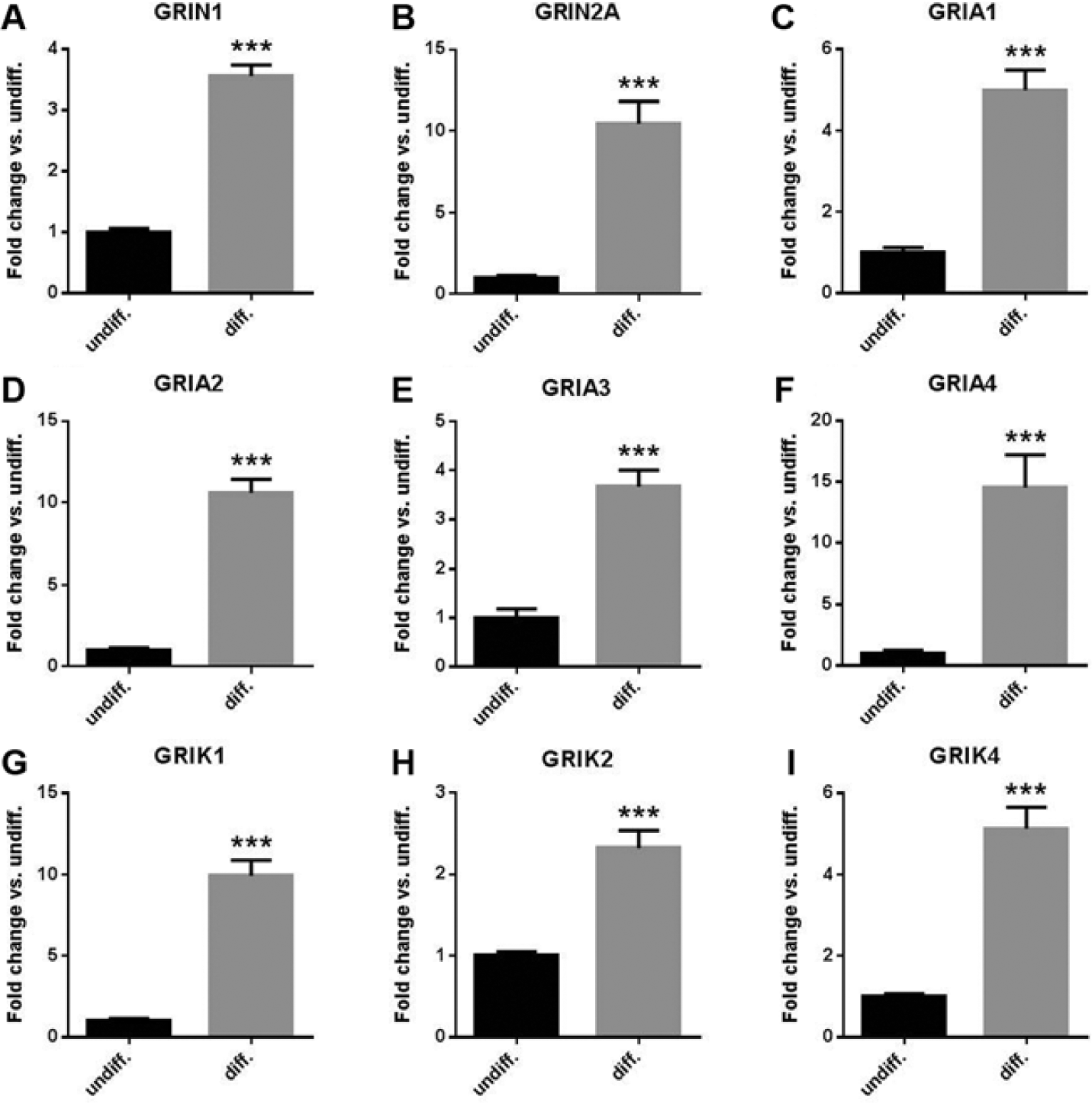
Messenger RNA (mRNA) expression of ionotropic glutamate receptors in HIP-009 cells. Quantitative reverse transcription PCR analysis showing upregulation of mRNA expression of N-methyl-D-aspartate receptors (NMDARs) GRIN1 (
Functional Analysis by Calcium Rise Assay of Ionotropic Glutamate Receptors Expressed in HIP-009 Cells
Upon stimulation with glutamate, NMDA, AMPA, or KA, no fluorescent signals were observed in undifferentiated HIP-009 cells (
Fig. 5A
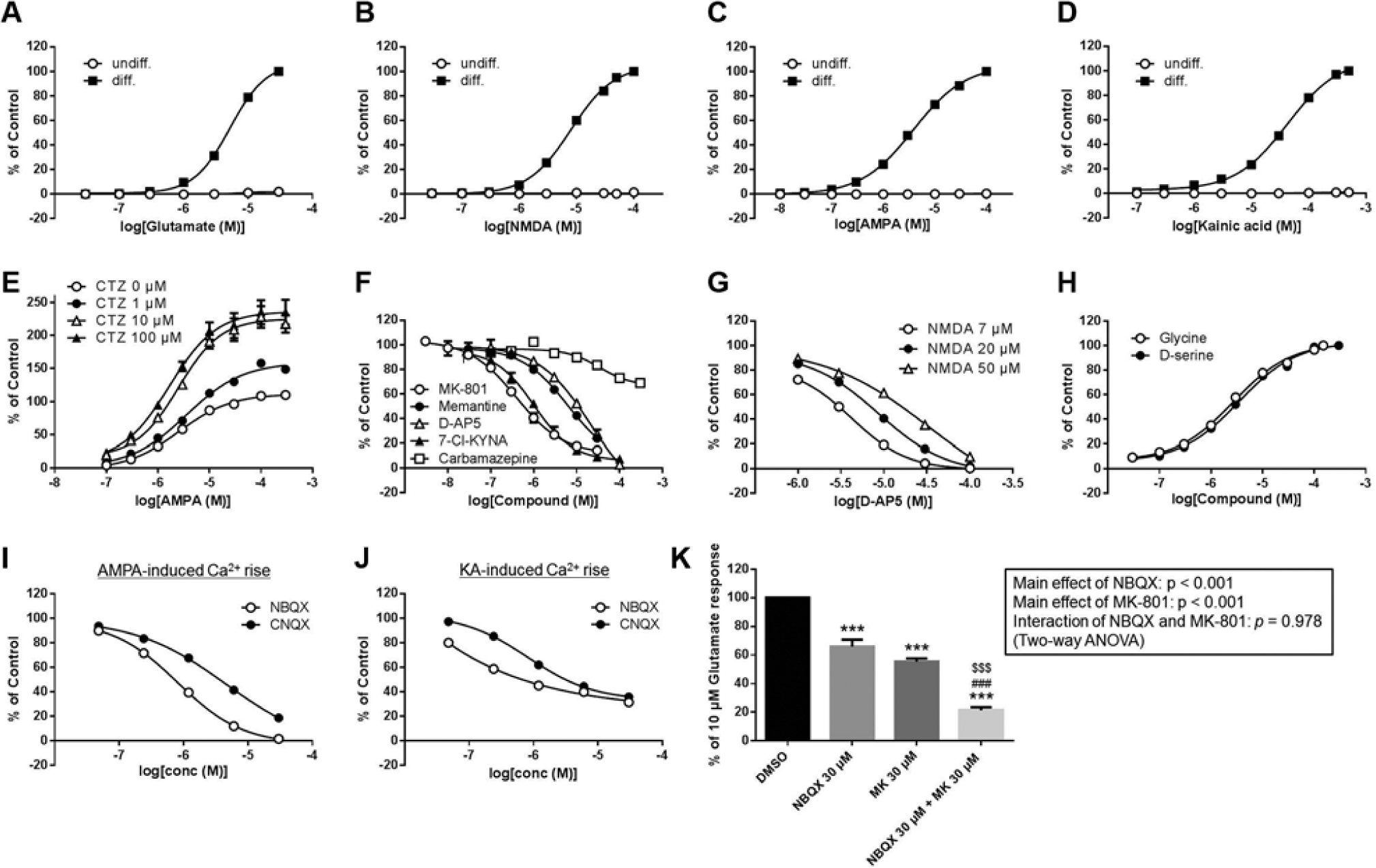
Calcium rise assays of ionotropic glutamate receptors expressing in HIP-009 cells. (
To further investigate the function of NMDARs in HIP-009 cells, we assessed the inhibitory effects of antagonists on NMDA-induced signals. MK-801, memantine, D-AP5, and 7-Cl-KYNA blocked the NMDA-evoked calcium rise in a concentration-dependent fashion ( Fig. 5F ). The IC50 values of each antagonist were as follows: MK-801, 0.6 ± 0.1 µM (n = 4); memantine, 6.7 ± 1.0 µM (n = 4); D-AP5, 11.4 ± 1.8 µM (n = 4); and 7-Cl-KYNA, 1.1 ± 0.2 µM (n = 3).Weak signal inhibition was observed with the antiepileptic drug carbamazepine; maximum (29.8% ± 2.4%) inhibition was observed at 300 µM (n = 4). The glutamate-site specificity of D-AP5 was confirmed by its concentration-dependent assay under various NMDA concentrations. An inhibition curve shift was observed in accordance with the change in NMDA concentration ( Fig. 5G ); the IC50 values of D-AP5 at 7, 20, and 50 µM NMDA were 2.8 ± 0.2, 5.4 ± 1.7, and 11.4 ± 1.8 µM, respectively (n = 3). To verify the glycine-site specificity of 7-Cl-KYNA, we conducted a competitive assay with a glycine-site agonist, glycine or D-serine. The NMDA-induced calcium rise blocked by 7-Cl-KYNA was recovered in a concentration-dependent manner by the addition of glycine or D-serine (EC50 = 2.1 ± 0.2 and 2.7 ± 0.3 µM for glycine and D-serine, respectively [n = 3]; Fig. 5H ).
Furthermore, we assessed the effects of NBQX and CNQX—antagonists of AMPARs and KARs—on the AMPA- or KA-evoked calcium rise to confirm the functions of these receptors in differentiated HIP-009 cells. NBQX and CNQX inhibited both AMPA- and KA-induced signals in a concentration-dependent fashion (NBQX, IC50 = 0.7 ± 0.1 and 0.7 ± 0.03 µM for AMPA and KA stimulation, respectively, n = 3; CNQX, IC50 = 3.8 ± 0.6 and 3.1 ± 0.4 µM for AMPA and KA stimulation, respectively, n = 3) ( Fig. 5I , J ). The AMPA-evoked calcium rise was completely inhibited by NBQX or CNQX ( Fig. 5I ), whereas 68.6% ± 1.3% or 64.2% ± 0.9% inhibition of the KA-induced signal was observed with 30 µM of NBQX or CNQX treatment, respectively (n = 3) ( Fig. 5J ).
To examine the degree to which each receptor contributed to the calcium rise upon glutamate stimulation, we tested the effect of co-treatment with MK-801 and NBQX on the glutamate-evoked calcium rise. When 30 µM of MK-801 or NBQX (at which concentration the NMDA- or AMPA-induced calcium rise was blocked completely) was added alone, 44.5% ± 2.2% or 34.2% ± 5.0%, respectively, of the glutamate-induced calcium rise was inhibited (n = 3) ( Fig. 5K ). Co-treatment had an additive inhibitory effect on total calcium rise upon glutamate stimulation (78.6% ± 2.2% inhibition; n = 3).
Discussion
In immunocytochemical and gene expression studies, we showed that undifferentiated HIP-009 cells had characteristics of neural stem/progenitor cells and that they were able to differentiate into neurons with gene expression profiles that closely resembled that of the human adult hippocampus (
Figs. 1
,
2
and
Here, we presented evidence for the first time that differentiated HIP-009 cells expressed functional NMDARs, AMPARs, and KARs. In the intracellular calcium rise assays, a concentration-dependent increase in intracellular calcium levels in response to an agonist of each receptor was observed in differentiated HIP-009 cells. Furthermore, each agonist-induced signal was inhibited by a selective antagonist of each receptor. Table 1 compares the EC50 or IC50 values of each agonist and antagonist for differentiated HIP-009 cells with data reported from rat hippocampal neurons.17–20 Some compounds had similar EC50 or IC50 values in the two species. However, compounds such as glutamate, AMPA, and NBQX exhibited one-order-smaller EC50 or IC50 values in differentiated HIP-009 cells than in rat hippocampal neurons. The differences in EC50 or IC50 values between differentiated HIP-009 cells and rat hippocampal neurons might reflect the species difference between humans and rats, although differences in assay conditions should also be taken into account. McNeish et al. 21 found in human and mouse ECS-derived neurons that some, but not all, potentiators of AMPARs that they tested acted on human and mouse AMPARs with different potencies. Therefore, HIP-009 cells could be used specifically to screen for drug candidates that are more effective in humans than in rodents, thus enabling the development of novel drugs. In addition, it would be possible that the cells are applied to mid- to high-throughput screening, because in our study, undifferentiated HIP-009 cells could be expanded up to passages 15 with consistent differentiation potency (data not shown): this is why we used HIP-009 cells before passages 15 (see Materials and Methods). Through 15 passages, large amount of cells required for assays would be generated.
Comparison of EC50 and IC50 Values for Each Compound from Differentiated HIP-009 Cells and Rat Hippocampal Neurons.

AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate; CBZ, carbamazepine; NMDA, N-methyl-D-aspartate.
Values are expressed as means ± SEM of three to four separate experiments unless otherwise indicated.
Data from Ambrósio et al. 17
Stimulated by
Stimulated by
Stimulated by
Stimulated by
Stimulated by
Stimulated by
Our results suggest that HIP-009 cells are a novel tool for functional assays of AMPARs and NMDARs under more physiologically relevant conditions than previously reported. Under physiological conditions, TARPs modulate extremely rapid desensitization of neuronal AMPARs.
6
Therefore, a desensitization inhibitor such as CTZ was needed to detect the activation of AMPARs in AMPAR-overexpressing cells without TARPs.6,22 Furthermore, McNeish et al.
21
found that human ESC–derived neurons were less sensitive to AMPA stimulation without CTZ. In contrast, HIP-009 cells were able to respond to AMPA alone, which was coincident with the results in rat primary hippocampal neurons.
23
This observation implies that not only AMPARs but also TARPs are expressed inherently in differentiated HIP-009 cells. Indeed, we observed upregulation of AMPARs and TARPs in differentiated HIP-009 cells at the gene expression level (
Fig. 4
and
It has been difficult to develop NMDAR assay systems using stably NMDAR-overexpressing cells without the use of conditional expression systems, such as tetracycline-inducible systems. 24 This is because of cytotoxicity caused by calcium influx via NMDARs upon prolonged activation by the agonist or agonists present in the culture medium (e.g., glutamate). By using HIP-009 cells, however, we were able to culture the cells and conduct functional assays of NMDARs without the need for such artificial expression systems. This finding suggested that some cytotoxicity-reducing mechanisms, such as glutamate uptake by astrocytes under physiological conditions, were present in differentiated HIP-009 cells. 25
In the intracellular calcium rise assays, we investigated in detail the functions of ionotropic glutamate receptors in differentiated HIP-009 cells. It is widely known that NMDARs require not only NMDA but also a coagonist, such as glycine or D-serine, to be activated. 26 In the calcium rise assay, however, we observed a concentration-dependent increase in intracellular calcium levels in response to NMDA alone in differentiated HIP-009 cells. This result suggests that coagonists are present in the differentiation medium used and remain in the assay buffer, even after several washes. Our observation that the signals upon NMDA stimulation were blocked by 7-Cl-KYNA supported this hypothesis. In functional assays for NMDARs, we tested binding site-specific antagonism by D-AP5 and 7-Cl-KYNA, which are, respectively, glutamate- and glycine-binding site–specific antagonists of NMDARs. 27 To confirm the glutamate-binding site specificity of D-AP5 in HIP-009 cells, we performed an inhibition curve shift assay under various NMDA concentrations. The curves were right-shifted with increasing concentrations of NMDA. This curve shift was not observed with other binding site–specific antagonists—namely, MK-801, memantine (both targeting ion channel sites), and 7-Cl-KYNA (targeting glycine-binding sites) (data not shown). Furthermore, we validated the glycine-binding site specificity of 7-Cl-KYNA in a competitive assay with a glycine-binding site agonist, glycine or D-serine.
By using NMDAR-functional assays, we also validated the contribution of carbamazepine—a well-known antiepileptic drug acting mainly by sodium channel blockade—to the NMDAR inhibition reported previously (43.1% ± 3.2% inhibition at 500 µM carbamazepine) in rat hippocampal neurons.
17
In agreement with the results of the previous report, the NMDA-induced calcium rise in differentiated HIP-009 cells was blocked by high concentrations of carbamazepine (29.8% ± 2.4% inhibition at 300 µM carbamazepine). Using rat hippocampal neurons, Ambrósio et al.
28
showed that carbamazepine inhibited L-type calcium channels that were activated subsequently to membrane depolarization through the activation of ionotropic glutamate receptors but that it did not affect the glutamate receptors directly. Thus, our result implies that differentiated HIP-009 cells also expressed L-type calcium channels functionally. We did detect upregulation of L-type calcium channels in differentiated HIP-009 cells at the gene expression level (
In addition, we investigated the contribution of each receptor to the calcium rise upon glutamate stimulation by co-treatment with MK-801 and NBQX. We estimated that ~45% of the total glutamate-evoked calcium rise was through NMDARs, and ~34% of the rise was through AMPARs and KARs. The remaining activity level of about 20% implied that there was a contribution from other glutamate receptors—that is, metabotropic glutamate receptors—although there would also be a contribution from residual KARs that were not inhibited completely by 30 µM NBQX.
Population analysis of differentiated HIP-009 cells by using neuron- and astrocyte-specific markers revealed that HIP-009 cells differentiated into either neurons or astrocytes in the same culture. This is another advantage of HIP-009 cells, because astrocytes are difficult to obtain simultaneously with neurons from the same human ESCs/iPSC sources, such as neural stem cells, by using currently available neural differentiation protocols. 30 There is a line of evidence that astrocytes play important roles in neuronal functions, including the functional maturation of neurons and synaptic plasticity.31,32 AMPARs/KARs expressed in astrocytes also play an important role in neuron-astrocyte communications: Schell et al. 33 showed that D-serine was enriched in astrocytes and that release of D-serine from the astrocytes was selectively enhanced by non-NMDA glutamate receptor agonists such as AMPA and KA. The astrocyte-derived D-serine then modulated neurotransmission through synaptic NMDARs. In addition, Fellin et al. 34 showed that glutamate released from astrocytes mediated neuronal synchrony by activating extrasynaptic NMDARs. Furthermore, some reports indicate that astrocytes are involved in glutamate excitotoxicity. Anderson and Swanson 25 reported that astrocytes were primarily responsible for glutamate uptake in the CNS and thereby influenced synaptic activity. In HD, reduction of glutamate uptake by astrocytes might be associated with excitotoxicity and abnormal production of neurotoxic molecules, leading to synaptic impairment. 35 Because differentiated HIP-009 cells are a mixture of neurons and astrocytes, they can be used to detect these important neuron-astrocyte interactions.
In conclusion, we found here that HIP-009 cells are a novel physiologically relevant tool for evaluating the effects of drug candidates on ionotropic glutamate receptors expressed in human hippocampal neurons surrounded by human astrocytes. This should help to develop novel therapeutic agents for glutamate receptor–related disorders such as AD, PD, HD, schizophrenia, and epilepsy.
Footnotes
Acknowledgements
We thank Dr. Mai Uesugi and Ms. Kayoko Ebihara for their technical advice and assistance. We are also grateful to Drs. Lee Dawson and Myles Fennell for their invaluable advice.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.