
Abstract
The nuclear receptor SET domain-containing family of proteins (NSD1, NSD2, and NSD3) is known to mono- and dimethylate lysine 36 of histone H3 (H3K36). Overexpression and translocation of NSDs have been widely implicated in a variety of diseases including cancers. Although the substrate specificity of NSDs has been a subject of many valuable studies, the activity of these proteins has never been fully characterized in vitro. In this study, we present full kinetic characterization of NSD1, NSD2, and NSD3 and provide robust in vitro assays suitable for screening these proteins in a 384-well format using nucleosome as a substrate. Through monitoring the changes in substrate specificity of a series of NSD constructs and using molecular modeling, we show that a basic post-SET extension common to all three NSDs (corresponding to residues 1209 to 1226 of NSD2) is essential for proper positioning on nucleosome substrates.
Introduction
Posttranslational modification of histones, including histone methylation, plays a significant role in the regulation of gene expression and has been implicated in a variety of diseases.1,2 Histone methylation is a heritable, but reversible, modification catalyzed by histone methyltransferases (HMTs), transferring a methyl group from S-adenosyl-L-methionine (SAM) to lysine (by protein lysine methyltransferases; PKMTs) or arginine residues (by protein arginine methyltransferases) on histone tails. Abnormal methylation of lysine 36 of histone H3 (H3K36) has been widely implicated in a variety of cancers.3,4 NSD1, NSD2, and NSD3 are three members of the nuclear receptor SET domain-containing (NSD) family of proteins that mono- and dimethylate lysine 36 of histone 3 (H3K36).5,6 These proteins are composed of similar domains (e.g., SET, plant homeo domain [PHD], and Pro-Trp-Trp-Pro motif [PWWP] domains), 7 some of which may contribute to chromatin binding (e.g., PHD5-C5HCH domain of NSD3). 8 There are reports indicating that NSD2 also methylates other histone residues. For example, NSD2 was reported to methylate H3K27, thereby contributing to the carcinogenesis of certain tumor types, including myeloma. Up-regulation of NSD2 in the blood cells of leukemia patients was also reported. 9 NSD2 has also been implicated in DNA damage response, regulating the methylation status of the histone H4 K20 residue and being involved in the recruitment of 53BP1 to sites of DNA damage. 10 Nimura et al. 11 originally proposed that H3K36 trimethylation by mouse NSD2, together with developmental transcription factors, functions to regulate transcription, preventing any dysregulation that may contribute to diseases. SETD2 has been reported to mediate H3K36 trimethylations. 12
Mutations and deletions of the NSD1 gene have been reported to cause Sotos syndrome, which is characterized by overgrowth, macrocephaly, distinctive facial features, and developmental delay. 13 The NSD2 gene, also known as WHSC1 (Wolf-Hirschhorn syndrome candidate 1), is located in the short arm of chromosome 4, called the WHS critical region (WHSC). A hemizygous deletion of the short arm of chromosome 4 and loss of the WHSC1 gene is considered to be responsible for the core phenotypes of this disease: facial dysmorphic features, microcephaly, prominent glabella, mental retardation, and pre- and postnatal growth delay. 14 Translocation of NSD genes and formation of fusion proteins that contribute to cancers have been reported for NSD1 (98 kDa nucleoporin [NUP98]–NSD1 fusion; acute myeloid leukemia [AML] 15 and myelodysplastic syndrome 16 ), NSD2 (immunoglobulin gene translocation; multiple myeloma 17 ), and NSD3 (NUP98-NSD3 fusion; AML 18 ). In 15% to 20% of multiple myeloma cases, the t(4;14) translocation fuses the NSD2 (multiple myeloma SET domain) gene to the immunoglobulin heavy-chain promoter (enhancer) and results in a dramatic increase in NSD2 expression. 19
Reversibility of epigenetic modifications provides an opportunity to counter the effects of overexpression of NSDs by identifying potent, selective, and cell-active inhibitors. In recent years, inhibitors of histone deacetylases (HDACs) and DNA methyltransferases have been approved for treatment of certain types of cancer.20,21 However, the screening efforts for NSDs have been hindered by the lack of optimized assays in plate formats. A comprehensive study by Reinberg’s lab showed that the target of NSD2 methylation depends on the nature of the substrates used in the in vitro assays. 5 Although SET domains of NSDs are active with nucleosome substrates, none of them are active if the H3K36 residue of the nucleosome is mutated to alanine. It was also reported that if a histone octamer is used as a substrate, histone H4 lysine 44 is the residue predominantly methylated by NSD2. 5 However, the mechanism of substrate specificity of NSDs and their interaction with nucleosomes are not well understood. Here we report on substrate specificity and full kinetic characterization of NSDs in vitro and identify a basic post-SET extension present in all three NSDs as an essential sequence for recognition of nucleosome as a substrate in vitro. We also provide robust in vitro assays suitable for screening these proteins in a 384-well format.
Materials and Methods
Cloning, Expression, and Purification of Human NSDs
DNA fragments encoding NSD1, NSD2, and NSD3 constructs were amplified by PCR and subcloned into the pET28a-MHL vector (Genbank ID EF456735), downstream of the poly-histidine coding region. All proteins were overexpressed in Escherichia coli BL21 (DE3) pRARE2-V2R by the addition of 1 mM isopropyl-1-thio-D-galactopyranoside and incubated overnight at 15 °C. Proteins were purified as described in the Supplementary Material and Methods.
Details of chicken nucleosome, histone octamer, and H3-H4 tetramer purifications are also presented in the Supplementary Material and Methods.
Enzymatic Activity
Methyltransferase activity was assessed using radioactivity assays that involve either the trichloroacetic acid (TCA) precipitation method or scintillation proximity method (SPA). When testing peptides as substrate, the SPA technique was used, in which the 3H-methyl group of 3H-SAM (~18 Ci/mmol; PerkinElmer, Waltham, MA) is transferred to biotinylated peptide substrates. In this assay, reactions in 10 µL volume were quenched by adding 20 µL guanidine HCl (5 M final) followed by 170 µL 20 mM Tris buffer, pH 8.0. The mixtures were then transferred to streptavidin-coated SPA plates (PerkinElmer), incubated for at least 1 h, and read with a TopCount radioactivity counter (PerkinElmer). When histone proteins were used as substrate, the TCA precipitation method was used where histones with incorporated 3H-methyl were precipitated with TCA. The 10 µL reaction mixtures were quenched by adding 90 µL of 10% TCA, transferred to a 96-well filter plate and dried under vacuum. The dried filters were washed two times with 100 µL of 10% TCA and once with 100 µL 100% ethanol, allowed to dry, followed by addition of 70 µL Microscint-0 solution (PerkinElmer) to each well. The CPM signal was read with a TopCount radioactivity counter (PerkinElmer). Reactions for assay development, assay optimization, and enzyme characterization were typically evaluated using 96-well MultiScreen HTS FB plates (Millipore Corp, Billerica, MA) and adapted to a 384-well format.
To assess the activity of different NSD enzymes with different substrates (histone peptides, histone octamers, H3-H4 tetramers, nucleosomes, or individual histones), activity assays were carried out in 50 mM Tris buffer, pH 9.0, and 0.01% Triton X-100 in the presence of 5 mM Tris (2-carboxyethyl) phosphine (TCEP) or 5 mM dithiothreitol (DTT). Each substrate was used at a final concentration of 5 µM. Activities were measured in quadruplicate by the TCA method. In the case of peptides, the SPA method was used.
For optimization of buffer conditions, the optimum pH was determined first by running the reaction in 50 mM Bis-Tris propane buffer, which allows a wide pH range of 6 to 9.5. Once the optimum pH was determined, the effect of other additives (salt, ions, reducing agents, detergent, etc.) was tested by systematically varying the studied parameter in the reaction mixture while keeping all other parameters constant. The optimized buffer condition was 50 mM Tris buffer, pH 9.0, 0.01% Triton X-100, 5 mM TCEP for all enzymes except for NSD2 (825-1208), where 5 µM dsDNA was added in addition to 200 mM NaCl, which came from the octamer preparation. Histone octamer complexes have low stability in the presence of low salt concentrations; therefore, they were kept at 2 M NaCl and diluted 10-fold into the reaction mix just before starting the reaction.
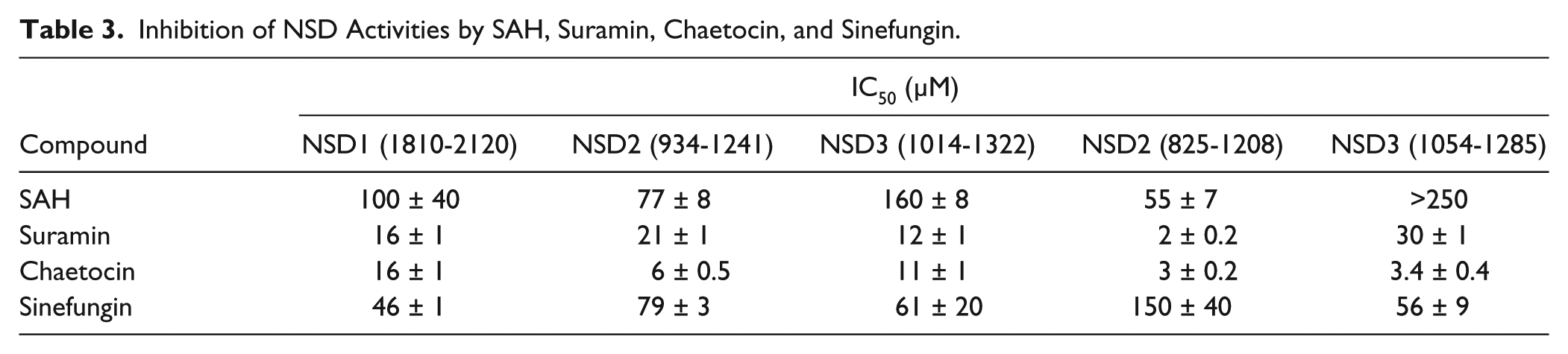
For Km and kcat determinations, the TCA precipitation assay was used to determine the initial velocities of the NSD enzyme catalyzed reactions in the presence of varied concentrations of substrate and SAM cofactor. An optimized buffer for each enzyme was used, and Km values for both substrates were determined by varying the concentration of one substrate while keeping the other at saturation. Km values were calculated by fitting initial velocities versus substrate concentrations using nonlinear regression (SigmaPlot software). For IC50 determinations, assays were performed at concentrations close to the Km of both substrates in the presence of varied amounts of the inhibitor, and data were fitted using the four-parameter logistic equation (SigmaPlot).
For Z′-factor determinations and screening, the TCA assay was adapted to a 384-well format using AcroPrep 384 filter plates (PALL Corp, Washington, NY). Reaction volumes were reduced to 5 µL and 4.5 µL of the reaction mixture, containing enzyme and substrate in assay buffer, and were dispensed into each well of an assay plate (V-bottom small-volume 384-well plate from Greiner, Monroe, NC) using the reagent dispenser Multidrop Combi (Thermo Scientific, Waltham, MA). Subsequently, using an Agilent Bravo automated liquid handler equipped with a 384-channel head (www.agilent.com), 250 nL of each compound solution was transferred to the 384-well assay plate and incubated, and were 250 nl 3H-SAM was added to start the reaction. Typically, a 5 µL reaction mixture contains ~75 nCi of 3H-SAM mixed with unlabeled SAM. All experiments were performed at 23 °C for 60 min using concentrations of both substrates around Km values. After incubation, reactions were quenched by adding 60 µL of 10% TCA, transferred to 384-well filter plates, and washed twice by TCA (60 µL) followed by one 60 µL ethanol wash. All washes were performed using the Bravo liquid handler and the Multiscreen vacuum manifold (Millipore). The plates were left to dry at 37 °C for at least 1 h, 20 µL of MicroScint-0 (PerkinElmer) was transferred to each well, and the CPM signal was read using the TopCount radioactivity counter (PerkinElmer). DMSO and 200 µM suramin were used as negative and positive controls, respectively.
Molecular Modeling
Molecular modeling was performed as described in the Supplementary Material and Methods.
Results
Many HMTs, including NSDs, are relatively large proteins (e.g., NSD1 consists of 2696 amino acids) and hard to express and purify in sufficient quantities for three-dimensional structure determination, screening efforts, or in vitro characterization. However, it has been shown that for many HMTs, constructs consisting of the core methyltransferase SET domain as well as adjacent pre-SET and post-SET domains are mostly active. We used the same approach to identify soluble, easy-to-purify, and active constructs of NSDs.
Identifying Active Constructs of NSDs
Multipurpose constructs were independently designed for NSD1, NSD2, and NSD3. Among them, NSD1 (1810-2120), NSD2 (825-1208), and NSD3 (1054-1285) were soluble enough and purified (>95% purity;
Fig. 1
). All three pure proteins were tested for activity using nucleosome, octamer, H3-H4 tetramer, and histone H3 residues 21 to 44 (H3 [21-44]) as substrates. Significant activity was observed for NSD1 (1810-2120) using nucleosome as a substrate (kcat: 92 ± 10 h−1, Km nucleosome: 0.21 ± 0.04 µM). However, this construct was not active with octamer, tetramer, or H3 (21-44) peptide substrates (
Table 1
). Surprisingly, NSD2 (825-1208) construct was not active with nucleosome as substrate, even though it is more than 65% identical to NSD1 (1810-2120) over 311 amino acids and consists of SET, pre-SET, and post-SET domains (
Table 1
;

Schematic representation of NSD constructs. NSD1, NSD2, and NSD3 constructs characterized in this study are represented. Each construct has been identified by the name of the protein followed by amino acid boundaries in brackets. The basic post-SET extension is represented in green in the alignments. The amino acid sequence corresponding to 1209 to 1226 of NSD2 is shown in red on the top. Blue arrows indicate the end of each truncated construct. PHD, plant-homeodomain; PWWP, Pro-Trp-Trp-Pro domain; AWS, associated with SET domain.
Kinetic Characterization of NSDs.
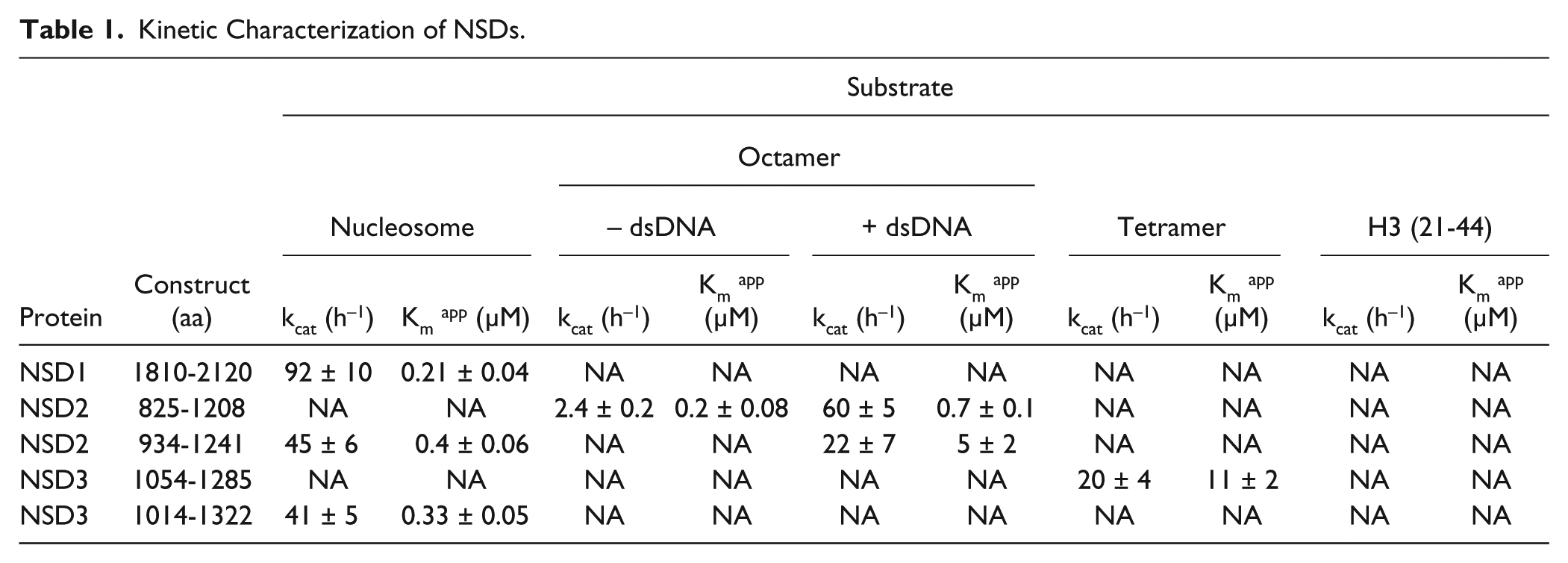
The activity of the NSD2 (825-1208) construct with octamer as substrate was stimulated in the presence of 41 base-pair double-stranded DNA (dsDNA;
New NSD2 and NSD3 Constructs That Can Methylate Nucleosome
The NSD1, NSD2, and NSD3 constructs showed differences in substrate specificity, despite their high amino acid sequence similarities. We hypothesized that the basic post-SET extension corresponding to amino acids 1209 to 1241 of NSD2 may play a role in conferring methylation activity against the nucleosome. We therefore cloned NSD2 (934-1241), that is close to the sequence used by Li et al.
5
for NSD2 (941-1240), and NSD3 (1014-1322) constructs that mimic the boundaries of nucleosome-methylating NSD1 construct (1810-2120;
Fig. 1
). As predicted, the new NSD2 and NSD3 constructs were active with nucleosome as substrate and their assay conditions were optimized in a similar manner as that for NSD1 (1810-2120;
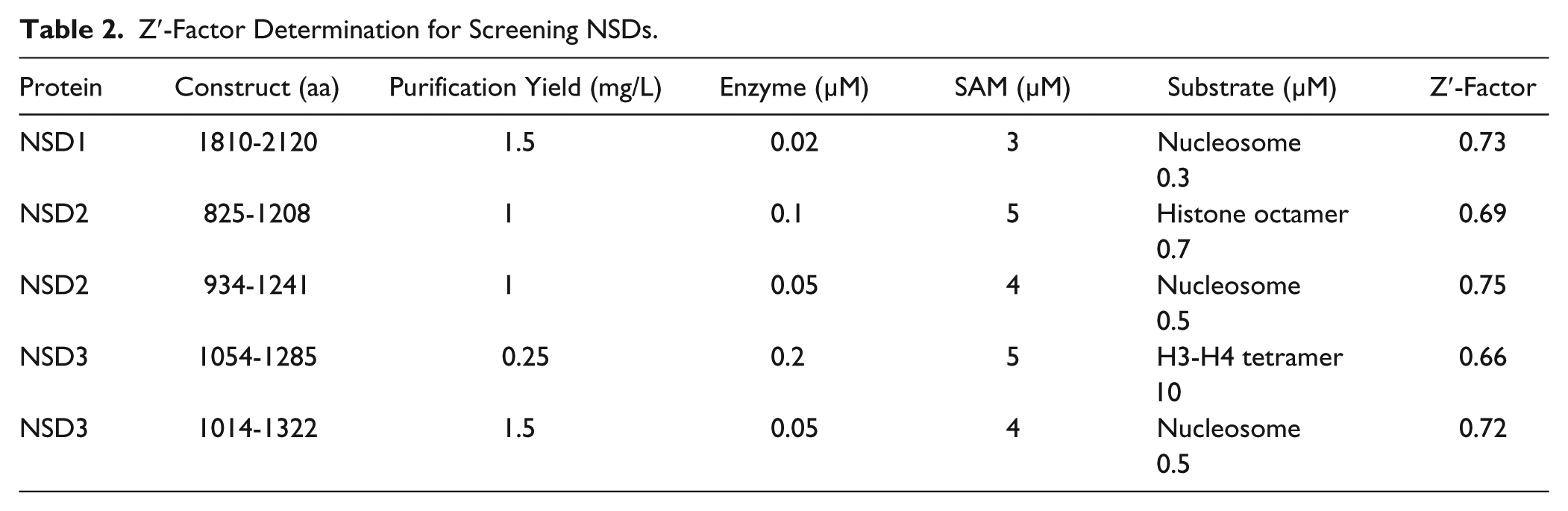
Enzyme assays for NSD1, NSD2, and NSD3 were further used for screening in 384-well format in low volume (5 µL) at Km of both substrates. In the presence of suramin as an inhibitor and nucleosome as a substrate, Z′-factor values
22
of 0.73, 0.75, and 0.72 were determined for screening NSD1 (1810-2120), NSD2 (934-1241), and NSD3 (1014-1322), respectively (
Table 2
;
Z′-Factor Determination for Screening NSDs.
Inhibition of NSD Activities by SAH, Suramin, Chaetocin, and Sinefungin.
Mapping the Basic Post-SET Extension
To identify the residues essential for the interaction of NSDs with nucleosome within the basic post-SET extension, we systematically deleted residues from the C-terminus of the NSD2 (934-1241) construct, generating seven new constructs of NSD2 ending at amino acid 1238, 1232, 1226, 1220, 1214, 1208, or 1202 (
Fig. 1
). All constructs were tested for activity using nucleosome as substrate. Deletion of C-terminal residues up to arginine 1226 had no effect on nucleosome methylation by NSD2 constructs, with NSD2 (934-1226) being fully active with nucleosome as substrate (kcat: 40 ± 1 h−1). However, all other constructs with additional deletions lost their ability to methylate nucleosome, indicating that residues 1221 to 1226 of NSD2 are essential for its interaction with the nucleosome (
Fig. 2
). We performed these assays in the presence and absence of the 14 bp dsDNA to test if the constructs missing 1221 to 1226 residues can regain activity with nucleosome as a substrate in the presence of additional dsDNA. No significant effect was observed for any of the tested constructs (
Fig. 2
). We further tested the effect of 14, 41, and 145 bp dsDNA on activity of NSD2 (934-1241), indicating the presence of dsDNA rather has a significant inhibitory effect at concentrations as low as 0.1 µM (
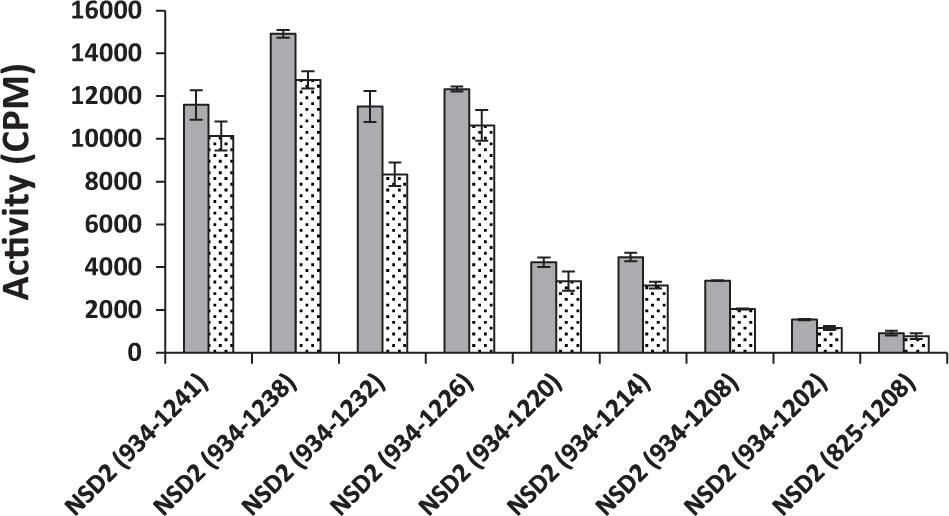
Mapping the basic post-SET extension. The activity of seven different truncated NSD2 constructs was assessed using nucleosome as substrate in the presence (dotted) and absence (solid) of 5 µM 14 bp dsDNA. NSD2 (934-1226) and longer constructs were fully active. However, shorter constructs lost the ability to methylate nucleosome. Experiments were performed in triplicate.
Modeling the Interaction of NSDs with Nucleosome
Unlike other common sites of histone posttranslational modification such as H3K9 or H3K4, H3K36 is positioned immediately after the histone 3 tail exits nucleosomal DNA ( Fig. 3A ). Recognition of methylated H3K36 by the PWWP domain of PSIP1 is mediated by direct binding to nucleosomal DNA,26,27 and similarly, it was proposed that NSD1 interacts with nucleosomal DNA. 28 In an attempt to understand the structural mechanism underlying increased nucleosome methylation driven by the C-terminal domain of NSD1, we built a model of NSD1 bound to nucleosome, starting from an experimental structure of the protein in complex with SAM 28 (see the Supplementary Materials and Methods section for details). We found that NSD1 can sit on the DNA double helix that is wrapped twice around histone octamers, with clear shape and electrostatic complementarity ( Fig. 3 ), in a conformation apparently similar to that of a previously reported docking model. 28 The histone-binding groove is at the DNA interface; the N-terminal H3 tail extends between the doubly-wrapped DNA helix (as observed in the crystal structure of human nucleosome 29 ) toward the histone-binding groove of NSD1, which results in positioning the epsilon nitrogen of H3K36 at the site of methyl transfer. We find that the basic post-SET extension of NSD1 is perfectly positioned to make extensive, stabilizing Van der Waals and electrostatic interactions with DNA. Conversely, in the absence of DNA, this electropositive domain is expected to inhibit interactions with basic histone tails. In summary, docking crystal structures of NSD1 and the human nucleosome places the substrate lysine at the catalytic site and the electropositive post-SET extension at the interface of the electronegative nucleosomal DNA. This could explain the increased nucleosomal methylation observed in the presence of the basic post-SET extension.
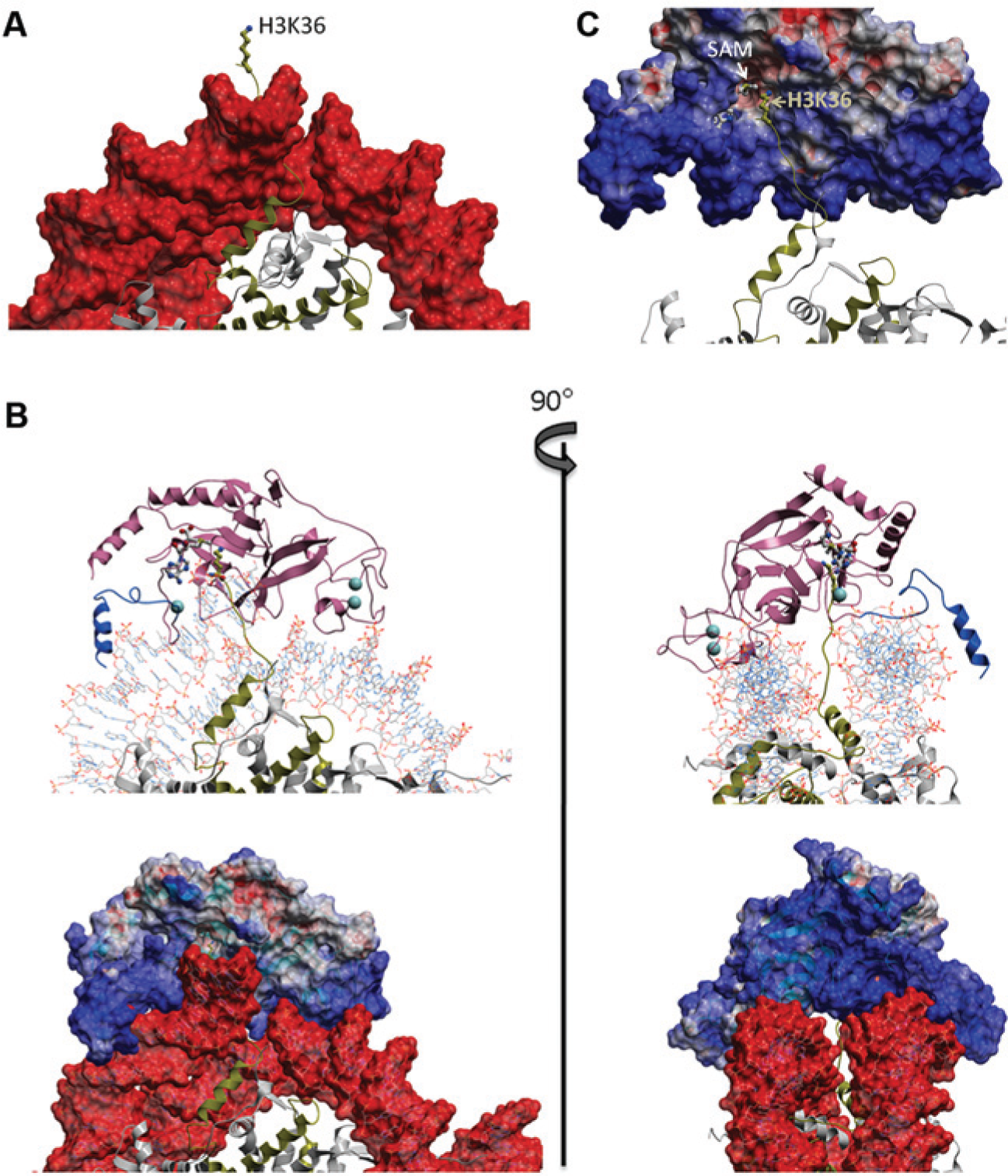
Model of NSD1 bound to nucleosome. (
Discussion
The substrate specificity of NSDs has been widely studied in vitro and in vivo. NSDs have been reported to methylate various histone lysine residues, including H3K4, 30 H3K27, 9 H3K36,5,6,11 and H4K20, 10 with biological functions associated with transcriptional activation or repression. After a comprehensive study, Reinberg’s group suggested that the methylation-mark specificity in vitro may change when using different substrates (e.g., histone peptides, core histones, or nucleosomes). 5 Reinberg showed that NSDs methylate H3K36 when DNA is present, either as a component of nucleosome or when it is simply added as small dsDNA fragments to histone octamers. In the absence of DNA, however, NSDs methylated H4K44 on octamer. No NSD2 activity was detected when H3K36 on nucleosome was mutated to alanine (H3K36A). In contrast, activity was detectable on octamers containing H3K36A mutation, an indication that when nonnucleosome substrates are used, other residues such as H3K4 and H3K27 may possibly be methylated. 5 Now it has been unambiguously clarified that NSDs are H3K36 dimethylases both in vitro and in vivo.5,6
The crystal structure of NSD1 shows that a post-SET loop adopts an autoinhibitory conformation that blocks the substrate-binding site. However, it was suggested that this inhibitory loop can adopt an active conformation that could be stabilized by nucleosome. 28 A similar pattern has been reported for ASH1L, an H3K36 methyltransferase, where the substrate binding pocket is blocked by a loop from the post-SET domain. 31 This may explain why NSDs are not active with peptides as substrates. In addition, a lack of robust quantitative assays has been a problem in kinetic characterization, miniaturization, and screening of NSDs. Because of the low level of activity of NSDs in vitro, methods such as fluorography, autoradiography, and Western blot have often been used to study their activities. Although these types of assays are valuable for qualitative studies, they are difficult to employ for kinetic characterization of proteins or high-throughput screening and often are associated with technical problems such as antibody quality. 32
In this study, we have developed sensitive radioactivity-based assays for all three NSDs with nucleosome as substrate, determined their kinetic parameters, and optimized the assays for high-throughput screening. Interestingly, the basic post-SET extension corresponding to residues 1209 to 1241 of NSD2 was essential for methyltransferase activity of all NSDs with nucleosome as substrate. The essential sequence was further narrowed down for NSD2 to residues 1221 to 1226 (TKKKTR), the deletion of which abolished the ability of NSD2 to methylate nucleosome. Based on our model, this region, which is highly enriched in basic side-chains, is perfectly positioned to make extensive, stabilizing interactions with DNA consistent with nucleosomal proximity of H3K36. Similarly, the C-terminal of human DOT1L has been shown to be spatially distant from the SAM binding site yet important for DOT1L activity with nucleosome. It was suggested that the positively charged c-terminal region of DOT1L is critical for its interaction with nucleosomal DNA. 33 Similar interactions may also exist for other histone methyltransferases, for which further investigation is needed.
Although the catalytic efficiencies of NSDs are not dramatically different in vitro, their overexpression is associated with distinct cancers, indicating that they have nonredundant functions.4,34 The presence of different chromatin-interacting domains, such as PHDs and PWWPs, could contribute to their functional specificity. 8 Other posttranslational modifications, such as H2A ubiquitination, have been reported to affect H3K36 methylation by NSDs. 35
In summary, we have identified NSD1, NSD2, and NSD3 constructs that are active with nucleosome as substrate in vitro, determined their kinetic parameters, optimized the assays for high-throughput screening, and identified a basic post-SET extension that is present in all NSDs and is essential for their interaction with nucleosome. These assays pave the way for large-scale screening efforts that may lead to identifying potent and selective inhibitors that can be used in discovering distinct and nonredundant functions of NSDs in vivo and development of potential cancer therapeutics.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada through the Ontario Genomics Institute (OGI-055), GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Research and Innovation, Pfizer, Takeda, and the Wellcome Trust (092809/Z/10/). C.H.A. holds a Canada Research Chair in structural genomics.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.