
Abstract
Bioactive small molecules are an invaluable source of therapeutics and chemical probes for exploring biological pathways. Yet, significant hurdles in drug discovery often come from lacking a comprehensive view of the target(s) for both early tool molecules and even late-stage drugs. To address this challenge, a method is provided that allows for assessing the interactions of small molecules with thousands of targets without any need to modify the small molecule of interest or attach any component to a surface. We describe size-exclusion chromatography for target identification (SEC-TID), a method for accurately and reproducibly detecting ligand-macromolecular interactions for small molecules targeting nucleic acid and several protein classes. We report the use of SEC-TID, with a library consisting of approximately 1000 purified proteins derived from the protein databank (PDB), to identify the efficacy targets tankyrase 1 and 2 for the Wnt inhibitor XAV939. In addition, we report novel interactions for the tumor-vascular disrupting agent vadimezan/ASA404 (interacting with farnesyl pyrophosphate synthase) and the diuretic mefruside (interacting with carbonic anhydrase XIII). We believe this method can dramatically enhance our understanding of the mechanism of action and potential liabilities for small molecules in drug discovery pipelines through comprehensive profiling of candidate druggable targets.
Keywords
Introduction
Forward chemical genetics is the interrogation of cellular signal transduction pathways through the use of small molecules. Typically in this approach, a cellular assay is screened with a chemical library and hits are identified as compounds producing a desired phenotype. This type of profiling is often referred to as black-box screening or phenotypic screening because of the lack of a global perspective for all cellular networks.
In pharmaceutical drug discovery projects, medicinal chemistry is often applied to advance hits from phenotypic screens into lead compounds that display desirable pharmacodynamics (PD) and pharmacokinetics (PK) without ever knowing the precise molecular mechanism. In some cases, such leads are advanced to clinical trials without any knowledge of the cellular efficacy target for the small-molecule drug. Levetiracetam and pemirolast are two examples of new molecular entities approved by the Food and Drug Administration (FDA) for which neither the target nor the molecular mechanism of action has been established. 1
Despite forward chemical genetics’ potential for identifying new lead molecules, not knowing the efficacy target for a candidate small-molecule drug can represent significant risks. One example is potential on-target toxicity that perhaps can be best addressed in genetic animal models prior to moving to a phase I clinical trial. In addition, knowledge of the molecular target can enable structure-based chemistry design to yield a high-affinity drug-target interaction and also the development of better biomarker assays to quantify compound PD. Furthermore, knowing the target can facilitate a target-focused screening campaign to identify novel chemistry with more desirable PK/PD properties and/or a different mechanism of action. Therefore, a forward chemical genetics strategy that embraces small-molecule target identification and validation can increase the number of novel target-lead pairs going into human proof-of-concept trials for the treatment of diseases for which either ineffective medicines or no therapy currently exists.
In 2004, Muckenschnabel et al. 2 reported the use of SpeedScreen technology, a 96-well format “sandwich”-based assay that uses size-exclusion chromatography (SEC) coupled to liquid chromatography (LC) and mass spectrometry (MS) to identify small molecules that interact with protein targets. In most affinity selection–mass spectrometry (ASMS) profiling techniques like SpeedScreen, the protein or target is specified and the objective is to find an interacting molecule(s) from a chemical library. We reasoned that this technology could be reworked whereby the protein or target is the variable and the small molecule is kept constant. Here we describe a platform for small-molecule target identification called SEC-TID: size-exclusion chromatography for target identification. To increase throughput, the original technology was modified and is now based on the use of size-exclusion chromatography in 384-well format for resolving small-molecule ligand-receptor interactions. In 384-well plates, small molecules are mixed with purified molecules, small molecule–target interactions are resolved by size-exclusion chromatography, and the amount of small molecule bound to target is quantified using LC coupled to MS. This method of profiling is not biased toward a target molecule class as we present examples of reliably detecting interactions of drugs with nucleic acids and several target protein classes. Because mass spectrometry is used to quantify the amount of small molecule interacting with targets, SEC-TID is not dependent on chemical modification of the compound for detection. In addition, we show that SEC-TID data, using either titrations of target or small molecule, can be used to measure binding affinity (Kd). Last, we describe the production and screening of a Protein Screening Library (PSLv1.0), a collection of approximately 1000 proteins representing diverse protein classes derived from the protein databank (PDB). We discuss the data from the profiling of staurosporine (a broad-spectrum kinase inhibitor), XAV939 3 (a selective poly [ADP-ribose] polymerase [PARP] inhibitor), mefruside 4 (a benzene-sulfonamide diuretic developed by Bayer (Leverkusen, Germany), and vadimezan 5 (a tumor vascular-disrupting agent). Given that in one platform, multiple target classes can be screened for interactions with small molecules, SEC-TID can contribute to developing new insights into both compound mechanism of action and preclinical safety.
Materials and Methods
Materials
Staurosporine was purchased from Enzo Life Sciences (Farmingdale, NY). Mefruside (4-chloro-N1-methyl-N1-[(2-methyltetrahydrofuran-2-yl)methyl]benzene-1,3-disulfonamide), ASA404 6 and XAV939, 3 +JQ1, 7 and bromosporine (www.thesgc.org/chemical-probes/bromosporine) were synthesized at Novartis as per published methods. Doxorubicin was purchased from EMD Millipore (Billerica, MA). COX-1 (bovine) and COX-2 (human recombinant) were purchased from Cayman Chemicals (Ann Arbor, MI). Carbonic anydrase XIII was purchased from R&D Systems (Minneapolis, MN). 4-Nitrophenyl acetate (4-NPA), 2,6-dichlorophenolindophenol sodium salt, coenzyme Q10, L-dihydroorotic acid, brequinar sodium hydrate, farnesyl pyrophosphate (FPP), geranyl pyrophosphate (GPP), and isopentenyl pyrophosphate (IPP) were purchased from Sigma (St. Louis, MO). Farnesyl S-thiolodiphosphate (FSPP) was purchased from Echelon Biosciences (Salt Lake City, UT).
Protein Screening Library (PSLv1.0)
In total, 10 µL of BL21-CODONPLUS (DE3)-RIL (Agilent Technologies, Santa Clara, CA) cells was seeded in wells of 96-well skirted PCR plates (cat. 82006-704; VWR, Radnor, PA) and incubated for 10 min with 1 µL of miniprepped DNA per well on ice. The plates were heat shocked for 45 s at 42 °C, then placed on ice for 2 min. Then, 25 µL of SOC media was added to each well and cells were left to recover for 1 h at 37 °C while shaking at 225 rpm. Next, 4 µL of the cultures was seeded on omnitray LB/Kan and LB/Carb agar plates (cat. L2025 and L2008, respectively; Teknova, Hollister, CA) using a Biomek FX (Beckman Coulter, Brea, CA). The plates were incubated overnight at 37 °C. Colonies were picked the following morning using a 96-well pronger (V&P Scientific, San Diego, CA) and transferred to 400-µL starter cultures of terrific broth (TB) with appropriate antibiotic in 96-well 1-mL blocks (cat. 3959; Costar-Corning, Tewksbury, MA). The starter cultures were grown overnight at 30 °C at 275 rpm. The starter cultures were used to inoculate eight copies of 1.3-mL cultures in 2-mL deep-well blocks (cat. 3960; Costar-Corning), 25 µL to a copy. The inoculated cultures were grown to an average OD of 0.4 at 37 °C, after which the temperature was dropped to 18 °C and all wells induced with 13 µL of 0.1M IPTG. Cultures were grown overnight, then pelleted by centrifugation in a tabletop centrifuge. During centrifugation, two blocks of culture were combined so a single bacterial pellet was obtained for every two blocks of culture grown. Pellets were either used immediately or frozen at −80 °C for later purification.
Bacterial pellets in 96-well blocks were allowed to thaw, if frozen, and suspended in 200 µL of Q-proteome lysis buffer (cat. 37900; Qiagen, Valencia, CA) per well, containing one Complete Protease tablet (cat. 05056489001; Roche, Indianapolis, IN) per 60 mL of lysis buffer. Pellets were resuspended by shaking with minimal vortexing if necessary. Resuspended pellets were shaken at 4 °C at 800 rpm for 30 min. Then, 400 µL of wash buffer I (50 mM Tris [pH 7.5], 500 mM NaCl, 10% glycerol, 15 mM imidazole) was added to each well, and the plates were centrifuged at 4000 rpm for 10 min at 4 °C to pellet cellular debris. Supernatants were transferred to 96-well 1-mL blocks (cat. 3959; Costar-Corning), and 100 µL of prewashed 33% Ni-NTA resin (cat. 1018240; Qiagen) slurry was added to each well. Plates were then sealed (Storage mate III, cat. 3080; Costar-Corning) and shaken at 4 °C for 1.5 h. During this incubation step, plates were inverted every 15 to 20 min to prevent sedimentation.
After incubation, plates were spun for 1 min at 2000 rpm. Then, 400 µL of supernatant was removed from the wells using a Biomek FX. The remaining slurry was then resuspended and transferred to 850-µL filter plates (cat. 5054; Pall Corporation (Port Washington, NY), under which 96-well polypropylene collection plates were placed. The filter-collection plate assemblies were then spun for 1 min at 1000 rpm to remove any remaining lysate. The resin in the filter plates was washed three times with 200 µL of wash buffer I. The wash steps were interspersed with 1-min spins as described above. Resin was then washed three times with wash buffer II (50 mM Tris [pH 7.5], 500 mM NaCl, 10% glycerol, 30 mM imidazole). After the last wash step, the plates were spun once more to remove any residual wash buffer and the collection plates were exchanged. Then, 65 µL of elution buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 10% glycerol, 250 mM imidazole) was added to each well. The resin was incubated with elution buffer for 1 h, after which the plates were spun as above, and the elution step was repeated. All elution fractions were then pooled and aliquots were saved for analysis.
Protein elutions were reformatted into 384-well plates (Greiner Bio-One, Monroe, NC) and subjected to analysis by capillary electrophoresis (Caliper GXII; PerkinElmer, Waltham, MA). Proteins were considered to have successfully expressed and purified when the major protein peak was detected within 25% of the expected molecular weight and concentration of the expected peak exceeded 100 nM. The median protein concentration of the Protein Screening Library (PSLv1.0) is approximately 8 µM with a median purity of approximately 89%. The vast majority of the library corresponds to human proteins (~83%) in addition to proteins from Cryptosporidium parvum, Leishmania major, Plasmodium berghei, Plasmodium falciparum, Plasmodium knowlesi, Plasmodium vivax, Plasmodium yoelii, Toxoplasma gondii, and Trypanosoma brucei.
SEC-TID Screening
To generate SEC plates with reproducibly accurate volumes of resin, both from well to well and from plate to plate, a one-piece aluminum 384-well resin loader was fabricated by computer numerical control milling. The loader resembles a 384-well plate, the wells of which hold 10 mm3 of dry resin, and has raised guides along three sides, designed to properly hold and align an inverted 384-well filter plate (cat. MZHVN0W10; EMD-Millipore, Billerica, MA). A schematic with complete specifications is provided (
The 384-well filter plates were loaded with 10 µL of Sephadex G50 DNA grade F resin (cat. 17-0573-01; GE Healthcare, Piscataway, NJ) per well, after which 55 µL of buffer A (50 mM HEPES, 50 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 0.01% Pluronic F127) was added to each well. Subsequently, another 10 µL of resin was added, followed by the addition of another 55 µL of buffer A. Plates were sealed and allowed to swell for an hour before use. When kept at 4 °C, plates are usable for up to a week.
Screening was done in duplicate for all PSLv1.0 screens. We used preplated sets of the library proteins in skirted 384-well PCR plates (cat. 82006-678; VWR), 6 µL per well. A mixture of screening compounds was made up at 2× concentration and 6 µL was transferred to each well of the assay plate. Assay plates were sealed and incubated at room temperature for 4 h. Premade SEC plates were allowed to warm to room temperature and were spun for 1 min at 900 g in a tabletop centrifuge (Eppendorf 5810R) just prior to use. The collection plate under the SEC plate was then changed to the assay collection plate (Matrix, Hudson, NH, 4312). Samples were transferred from the assay plate using a Biomek FX (Beckman Coulter) equipped with a 384-well head. The sample volume transferred was 10 µL, and the dispensing height over the SEC plate was set to 9.5 mm from the well bottom. SEC assemblies (SEC plate and collection plate) were then spun for 2 min at 900 g. SEC plates were discarded and collection plates were prepped for LC-MS detection. Then, 1 µM final concentration of internal standard compound and 0.5 mM sodium dodecyl sulfate (SDS) were added to each well. Total volume in each well was 40 µL.
LC-MS/MS Analysis
Compounds were analyzed using an Agilent 1100 binary pump (Agilent Technologies) coupled to a CTC HTC pal auto-sampler (Leap Technologies, Carrboro, CA) and either a Quattro Premier mass spectrometer (Waters, Milford, MA) or a TSQ Quantum Ultra Triple mass spectrometer (Thermo, Waltham, MA) (
Nuclear Magnetic Resonance Spectroscopy
Human farnesyl pyrophosphate synthase (FPPS), amino acids 6 to 353, was prepared in Escherichia coli as previously described. 8 15N,2H-TROSY experiments were carried out at 296K in 3-mm shigemi tubes using 15N,2H-isotope–labeled FPPS at a concentration of 9 mg/mL in 25 mM Tris (pH 8.0), 25 mM NaCl, and 2 mM dithiothreitol (DTT). Ninety-six scans and 198 increments per spectrum were acquired in a Bruker (Billerica, MA) AV800 spectrometer at an 800-MHz proton frequency.
Isothermal Titration Calorimetry
Quantitative binding of ASA404 to either wild-type (PDB ID: 3N6K) or mutant (T210S, PDB ID: 2QIS) FPPS was determined by isothermal titration calorimetry (ITC) using a GE MicroCal Auto-ITC200 (GE Healthcare). For both wild-type and mutant FPPS experiments, the syringe concentration (ASA404) was 340 µM (120 µL total syringe volume), and the cell concentration (FPPS) was 34 µM (400 µL total cell volume). For wild-type FPPS, runs were performed with 20 injections at 2 µL per injection. For mutant FPPS, runs were performed with 16 injections at 2.5 µL per injection. Wild-type FPPS was prepared in 10 mM Tris (pH 7.4), 25 mM NaCl, and 5 mM MgCl2. Mutant FPPS was prepared in 25 mM HEPES (pH 7.5), 150 mM NaCl, and 10% glycerol. FPPS proteins in either buffer were then diluted with DMSO to 3.4% final DMSO as solvent correction for ASA404. ASA404 was prepared by dilution from 100% DMSO stocks in the corresponding FPPS buffer. Raw data were curve fitted in Origin 7 (GE Healthcare) using the one-site fitting model.
Dynamic Scanning Fluorimetry
Protein was diluted in a dynamic scanning fluorimetry (DSF) buffer (50 mM Tris [pH 7.5], 50 mM NaCl) to 10 µM. Compound or equivalent amount of DMSO was diluted in DSF buffer to 200 µM. Then, 5 µL of protein was mixed with 5 µL of compound in a white 384-well PCR plate (cat. 04 729 749 001; Roche). Next, 2 µL of 50× SYPRO orange protein gel stain (S-6650; Invitrogen, Carlsbad, CA) diluted in DSF buffer was added to each well, and the plate was sealed with a clear seal (cat. MSB1001; Bio-Rad, Hercules, CA). The plate was vortexed, centrifuged at 3000 g for 1 min, and then placed in a Bio-Rad C1000 thermal cycler. A temperature gradient from 25 °C to 80 °C with steps every 0.5 °C, 30 s at each temperature was performed. The first derivative of fluorescence intensity is plotted versus temperature.
X-Ray Crystallography
Crystallization
Crystals of human FPPS (6-353) were grown at 19 °C from 1.2M sodium potassium phosphate (pH 4.7), 25% glycerol, by the vapor diffusion hanging drop technique. Protein stock was 16.7 mg/mL FPPS in 10 mM Tris (pH 7.4), 25 mM NaCl, 5 mM MgCl2 with 1.25 mM ASA404, and 2.25% DMSO. To achieve full occupancy, one crystal was transferred into a fresh drop of 1.2M sodium potassium phosphate (pH 5.3), 25% glycerol, containing 1.25 mM ASA404 and 2.25% DMSO and incubated overnight.
Data collection and structure determination
The crystal was flash-frozen into liquid nitrogen. X-ray data were collected at the Swiss Light Source (Villigen, Switzerland), beamline X10SA, with a Pilatus pixel detector and X-rays of 1.0000 Å wavelength. In total, 720 images of 0.25-degree oscillation each were recorded at a crystal-to-detector distance of 340 mm and processed with XDS. 9 The structure was determined by difference Fourier, the ligand was built into clear difference electron density in Coot, and the structure was refined with AutoBuster 2.11.2 (Global Phasing Limited, Cambridge, United Kingdom).
Biochemical Assays
Dihydroorotate dehydrogenase (DHODH) activity was measured using a colorimetric assay. Briefly, 190 µL of 25 nM DHODH in 100 mM Tris (pH 8.0), 50 mM NaCl, 0.05% Triton X-100, and 75 µM 2,6-dichlorophenolindophenol sodium salt was mixed with 5 µL of DMSO/compounds and incubated at room temperature for 15 min. Thereafter, 5 µL of 20 mM L-dihydroorotic acid (500 µM final) was added and mixed. Absorbance at 600 nM was measured at 25 °C. FPPS inhibition assay was carried out by incubating 0.5 nM FPPS, 3.12 µM GPP, 3.12 µM IPP, and test compound in assay buffer (20 mM HEPES [pH 7.5], 5 mM MgCl2, and 1 mM CaCl2 containing 5% DMSO). The enzymatic reaction was started by mixing 10 µL FPPS (1 nM) in assay buffer, 1 µL of the test compound at various concentrations, and 9 µL solution of GPP and IPP (6.94 µM each) in assay buffer. The series dilution of the test compound was prepared by 1:3 dilution with DMSO, with the highest concentration for the series being 10 mM. After a 60-min incubation at 25 °C, the reaction was quenched by adding 60 µL methanol containing 3.33 µM FSPP as internal standard for MS quantification. After the reaction was quenched, the samples were centrifuged at 10,000 g for 5 min. The supernatants were then injected into the LC-MS/MS system. The detection of FPP was carried out on a Quantum Ultra triple quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA) with a CTC auto sampler (Thermo Fisher Scientific) and HP1200 binary pump (Agilent Technologies). The separations were performed on a BetaBasic 4 Javelin Guards column (5 µm, 20 × 2 mm; Thermo Electron Corporation, Madison, WI). The column was eluted by gradient elution with solvent A (25 mM ammonium carbonate with 0.1% ammonium hydroxide, pH 9.0) and solvent B (methanol) at a flow rate of 0.8 mL/min. The gradient was started at 5% B, held for 0.25 min, stepped to 35% of B, held for 5.5 min, stepped back to 5% B, and held for 0.25 min. The multiple-reaction monitoring (MRM) transitions monitored in negative mode were 381.1 to 79.0 for FPP and 397.2 to 159.1 for FSPP at a collision energy of 42eV and 22eV, respectively. The tube lens for MS detection was set at 140ev. The CAXIII assay was performed by adding 90 µL of 1.65 µM CAXIII in 50 mM Tris (pH 7.5), 75 mM NaCl was mixed with 2 µL of 1 mM mefruside or topiramate in DMSO and preincubated at room temperature prior to addition of 10 µL of 5 mM 4-NPA in 10% glycerol. Activity was measured by monitoring absorbance at 400 nM with 60-s intervals.
Results and Discussion
384-Well Based Size-Exclusion/LC-MS
The basis of the current SEC-TID assay is a 384-well microplate-based SEC assembly (
The SEC-TID workflow involves four steps. The first step, binding, involves the incubation of a small molecule with a candidate set of targets in a 384-well plate. In the second step, the compound-target solutions are transferred to the SEC plate using liquid handling and dispensed directly on top of the G50 size exclusion resin. Subsequently, the assembly is centrifuged at 900 g for 2 min. During this period, the small molecule that is unbound to a target molecule does not elute into the collection plate because the small molecule enters the pores in the G50 resin and is retained. In contrast, a small molecule that binds a larger target molecule coelutes in the void volume. In step 3, the samples are injected into an LC-MS system using a method specific for the small molecule of interest. In the last step, hits that interact with the small molecule are identified as targets.
Given that many small-molecule drugs target nucleic acid or a protein, we first investigated if selective interactions of small molecules with proteins and nucleic acids could be detected. To do so, we chose two molecules for this study. Staurosporine 10 is a broad-spectrum adenosine triphosphate (ATP)–competitive protein kinase inhibitor. Doxorubicin 11 binds DNA and alters DNA replication. After mixing these molecules with DNA or a protein kinase (CDK16) and performing SEC-TID, we observe that doxorubicin interacts with DNA and not with CDK16 ( Fig. 1A ). Staurosporine selectively binds CDK16 ( Fig. 1B ). Thus, selective interactions of small molecules with both proteins and nucleic acid can be detected using SEC-TID.
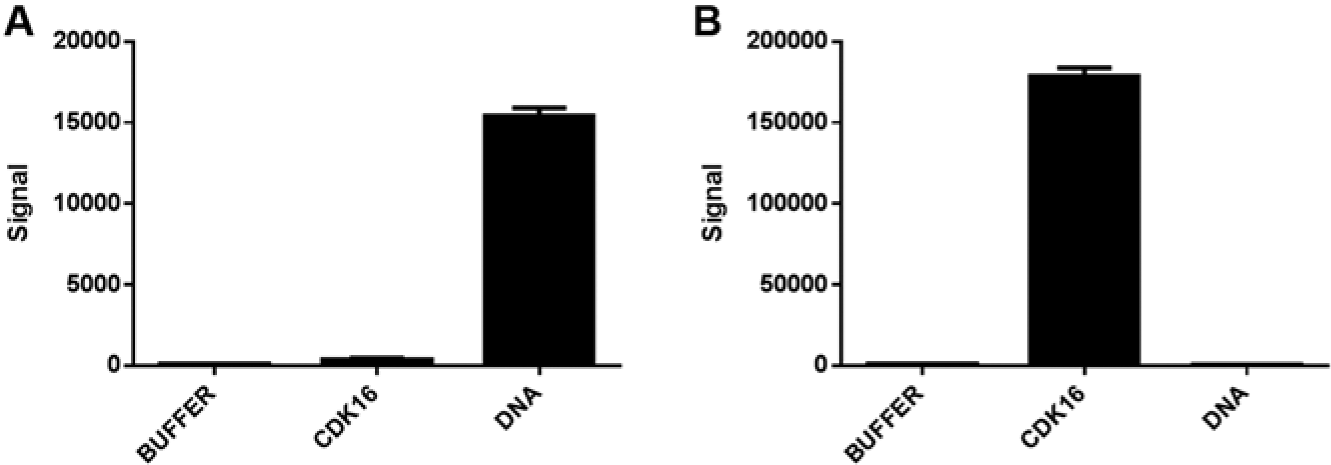
Nucleic acid and protein small-molecule interactions. Binding of doxorubicin (
Bromodomain Profiling with +JQ 1 and Bromosporine
Bromodomains are the only known protein domains that recognize and bind ϵ-N-acetylated lysine residues. 12 The human proteome has less than 50 proteins that contain this domain, and recently, selective small-molecule chemical probes have been identified that target these domains. To comparatively assess the binding profiles of known bromodomain interacting small molecules, we expressed and purified a panel of 31 bromodomain proteins, profiled two small molecules of interest, and evaluated the data relative to data obtained from DSF. DSF is a technique that can infer interactions of small molecules with proteins by measuring compound-induced changes in the thermal stability, or melting temperature (Tm), of a protein. 13
Filippakopoulos et al.
7
first reported that +JQ1 interacts with the bromodomains from BRD2, BRD3, BRD4, and BRDT. We profiled +JQ1 in SEC-TID and observed selective interactions with the bromodomains BRD2(1), BRD2(2), BRD3(1), BRD3(2), BRD4(1), and BRDT(1) (
Fig. 2A
). In addition, weak interactions were observed with BAZ2B and EP300. +JQ1 did not interact with any of the other 23 bromodomains. The same interactions of BRD2(1), BRD2(2), BRD3(1), BRD3(2), BRD4(1), and BRDT(1) with +JQ1 were observed using DSF (
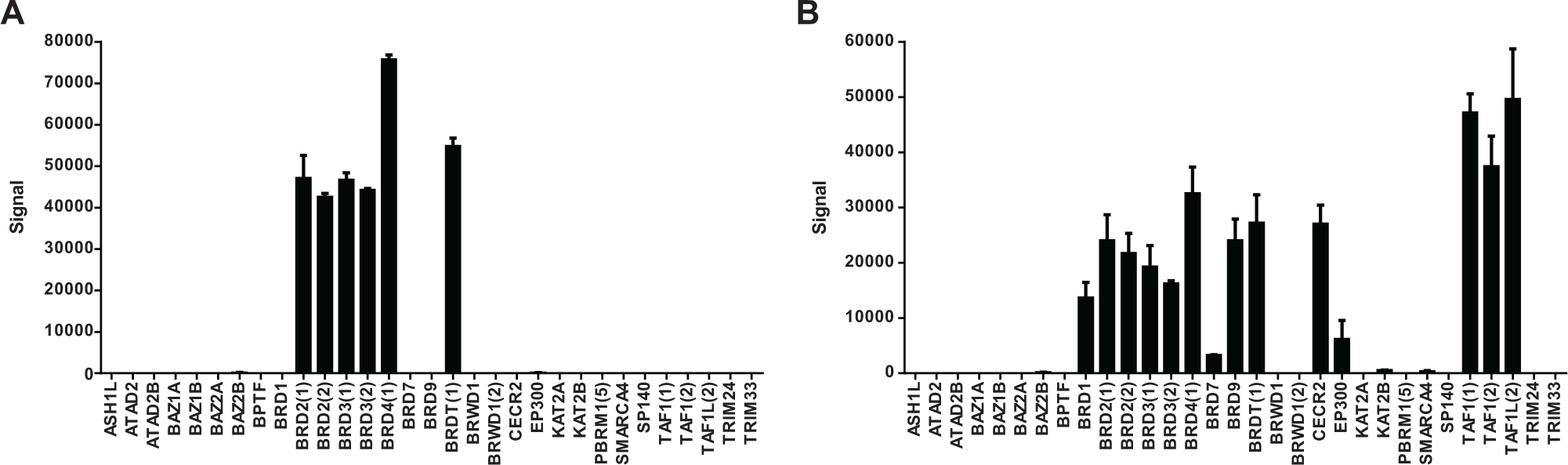
+JQ1 and bromosporine profiling of the bromodomain panel. Thirty-two bromodomains were expressed/purified from Escherichia coli and profiled by size-exclusion chromatography for target identification (SEC-TID). (
Quantitative Assessment of Small-Molecule Interactions
The ability to calculate an equilibrium dissociation constant (Kd) value from a SEC-TID experiment requires quantitation of the amount of small molecule bound to protein in each well. By adding a small-molecule titration to a dedicated section of each collection plate after size exclusion and the generation a standard curve, the amount of small molecule bound to protein can be extrapolated from the standard curve. After doing so, plots of the small-molecule concentration, [ligand-receptor] versus protein [receptor] or small-molecule [ligand] concentration can be analyzed to quantify a Kd. We first tested this using the enzyme dihydroorotate dehydrogenase (DHODH), an enzyme important for pyrimidine biosynthesis. 14 Using a DHODH biochemical assay with brequinar, a known potent DHODH inhibitor, an IC50 of approximately 10 nM is observed (data not shown). With 25 nM DHODH and titration of brequinar, an observed Kd of 18 nM is calculated ( Fig. 3A ). The true Kd is indeed lower than 18 nM but cannot be measured accurately given that 25 nM DHODH was required because of the limited sensitivity of the used LC-MS method for brequinar. As a second example, the interactions of a known inhibitor (diclofenac) for the prostaglandin-endoperoxide synthases 1 (COX-1) and 2 (COX-2) were quantified. Diclofenac 15 (a nonsteroidal anti-inflammatory drug) inhibits both COX-1 and COX-2 16 with IC50 values of 290 nM and 310 nM, respectively (data not shown). Using a fixed concentration of diclofenac and titrations of either COX-1 or COX-2, we observed similar results using SEC-TID ( Fig. 3B ). That is, diclofenac shows binding to both COX-1 and COX-2, with Kd values of 54 nM and 117 nM, respectively. To date, we have measured interactions of small molecules with targets having Kd values ranging from low nanomolar to approximately 50 µM. We have yet to address empirically the lower limit of affinity that can be detected in SEC-TID. We speculate the number is likely in the high micromolar range given that size exclusion is likely to be rapid (seconds) and considering complex half-time for a small molecule–target interaction with a dissociation rate of 0.1 s−1 (Kd of 100 µM when the association rate equals 103 M−1s−1) is approximately 7 s.
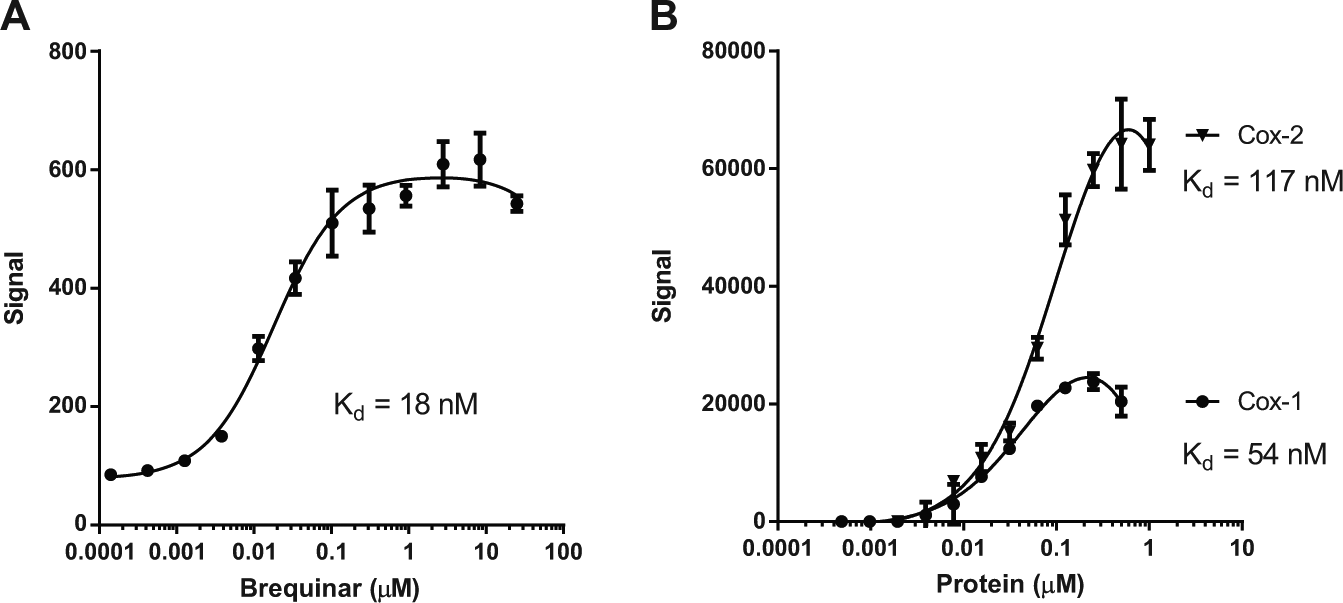
Quantification of affinity for receptor-ligand interactions. (
Profiling of Bioactive Compounds
To identify the efficacy targets of small molecules using SEC-TID, we reasoned that a high-quality protein library is needed. Therefore, we reached out to the SGC (www.thesgc.org) and acquired their protein expression constructs corresponding to proteins that have yielded protein structures. Only constructs designed for E. coli expression and purification were used. These constructs were transformed into an E. coli expression system, and the proteins were purified using metal-based affinity chromatography. The resulting protein library corresponds to proteins associated with 961 different PDB IDs and was named the Protein Screening Library (PSLv1.0).
To ask if we could identify known targets of small molecules using PSLv1.0, we first profiled staurosporine at a concentration of 10 µM. We observed 41 hits, and not surprisingly, all were protein kinases (
Fig. 4
and
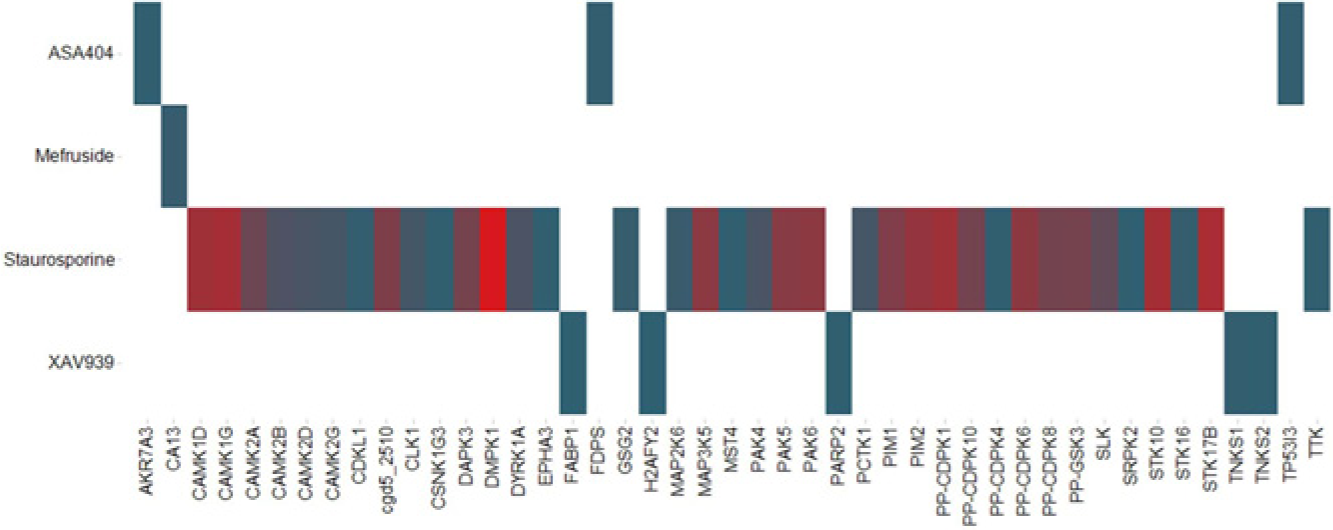
Compound profiling against the Protein Screening Library (PSLv1.0). Four small molecules—vadimezan/ASA404, mefruside, staurosporine, and XAV939—were profiled at a 10-µM concentration for interactions with PSLv1.0. A heat map of the raw data is shown. The y-axis lists the compounds, and the x-axis lists the targets (Gene Symbols) in the protein library. Values for Average(Signal) are represented by color, from low (blue) to high (red). The heat map illustrates a selectivity profile, and the absolute values are not to be compared across compounds because of the differences in detection sensitivity for each compound.
In a search to identify novel regulators of the Wnt pathway, Huang et al.
3
reported that XAV939 blocks Wnt signaling by inhibiting the NAD-dependent poly ADP-ribosylating enzymes tankyrase 1 (Tnks1) and tankyrase 2 (Tnks2). Because XAV939 binds to the active site within the PARP domain of both Tnks1
17
and Tnks2
18
and given that both Tnks1 and Tnks2 PARP domains are in PSLv1.0, we profiled XAV939 at 10 µM and observed five hits (
Fig. 4
and
Mefruside, a benzene-sulfonamide diuretic, is used for the treatment of hypertension and edema, although its molecular target has not been reported.
4
We screened mefruside at 10 µM and observed one hit, carbonic anhydrase XIII (CAXIII) (
Fig. 4
and
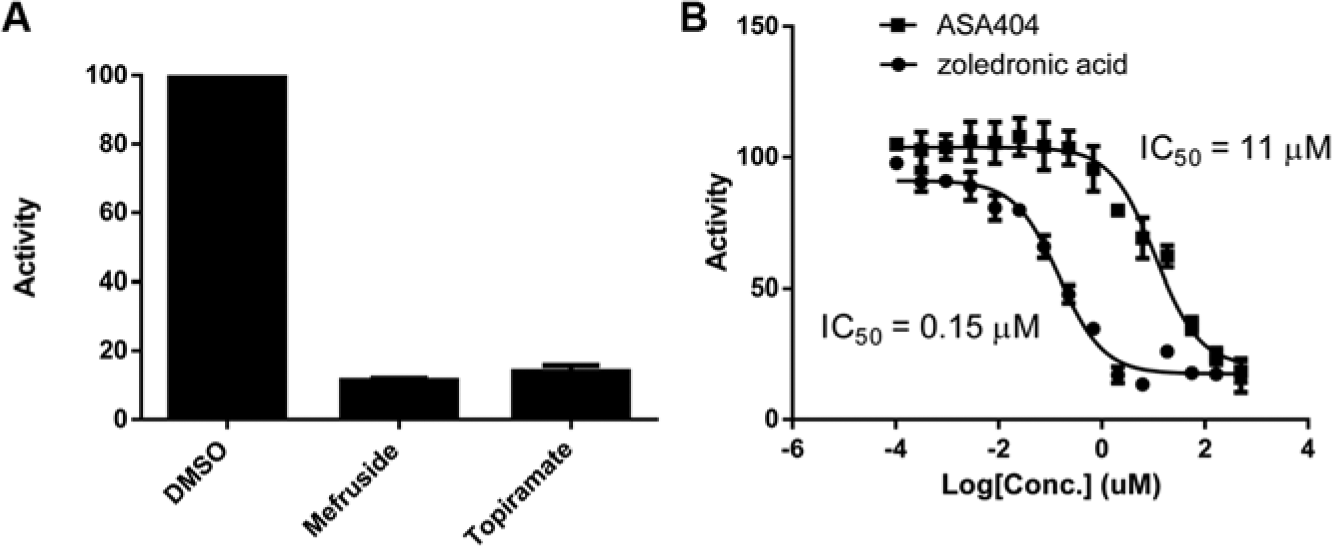
Validation of hits from compound profiling. (
Flavone acetic acid (FAA) is a compound that was identified to have broad antitumor activity in mice against mouse and human solid tumors. 23 Vadimezan/ASA404, an improved version of FAA, is an agonist for mouse STING 24 and activates the TBK1-IRF3 pathway 25 in macrophages, leading to the secretion of interferon-β (IFN-β). ASA404 binds to mouse STING with 130 nM affinity, yet no binding to human STING was observed even though human STING shares approximately 90% sequence identity to mouse STING. 25 Interestingly, ASA404 failed in phase III clinical trials, 26 and several reports24–25,27 speculate that this is due to the lack of binding of ASA404 to human STING.
Since it was reported that vadimezan/ASA404 has activity in human cells,
5
we sought to better understand the molecular targets of ASA404 by profiling ASA404 at a 10-µM concentration in SEC-TID. Interactions were observed with only three proteins: AKR7A3A, TP53I3, and FPPS (
Fig. 4
and
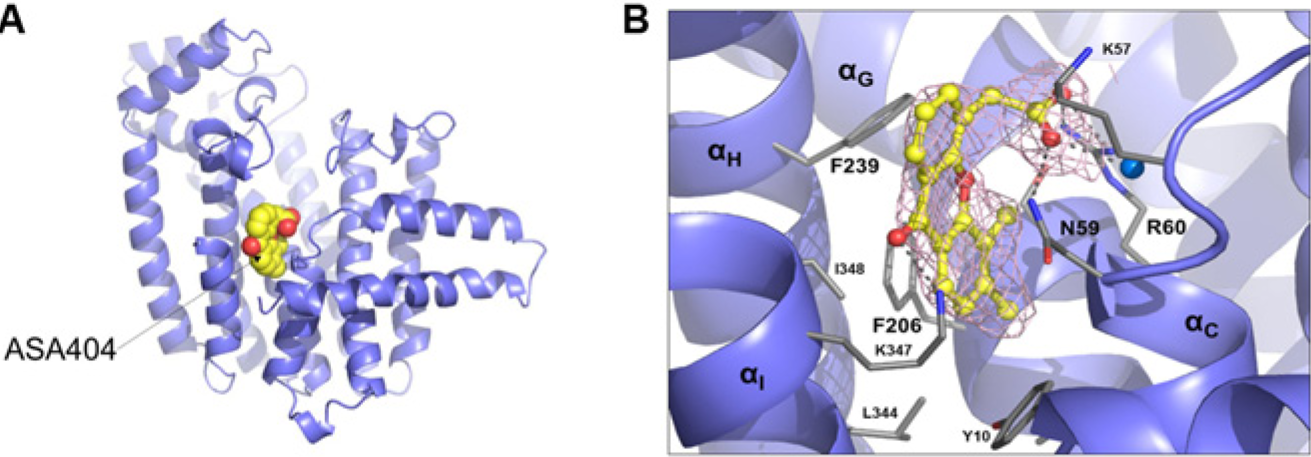
X-ray crystal structure for ASA404/farnesyl pyrophosphate synthase (FPPS) complex. (
SEC-TID is a reliable platform for measuring small-molecule target interactions, as expected interactions were observed for doxorubicin, staurosporine, +JQ1, bromosporine, XAV939, and diclofenac. SEC-TID can also be used to quantify the affinity of a small molecule for a target by performing either ligand or receptor titrations. We propose that the method described in this article for assessing small-molecule target interactions by LC-MS is superior to the method described by Arai et al. 31 for two main reasons. First, we have generated a protocol that allows for the use of very low protein concentrations, which expands the range of affinity that can be quantified. Second, after SEC, a low concentration of SDS is added to every well to break the compound-target association. By doing so, the addition of SDS increases the sensitivity of the assay. Because the methods described in this article allow for the use of very low protein concentration and addresses disassociation of the compound with the target prior to LC-MS, our method directly assesses the interaction of a small molecule with a target for calculating an affinity. As a proof that novel small molecule–protein interactions can be rapidly identified and validated, we chose a small set of bioactive small molecules and profiled them for interactions with a protein library consisting of approximately 1000 proteins.
We believe the results from XAV939 profiling represent a good example of the potential of SEC-TID for small-molecule target identification. Tnks1 and Tnks2, the known XAV939 cellular efficacy targets for modulation of Wnt pathway signaling, were identified as the top two hits in SEC-TID in addition to PARP2, an already known XAV939 interacting protein. In addition, two other interactions were observed with H2AFY2 and FABP1, which may represent off-target interactions. Although the interaction of XAV939 with the macro domain of H2AFY2 is likely of low affinity, it is of interest from the perspective that it is the only molecule other than metabolites, such as ADPR, ADP, AMP, or ATP, demonstrated to interact with this domain. 21 Thus, XAV939 may have value in generating a tool compound for probing H2AF and H2AFY2 cellular function.
Mefruside is a diuretic developed by Bayer, sold under the name Baycaron, for the treatment of edema and hypertension. 32 Mefruside hit only one protein, CAXIII, in our protein library. CAXIII is a novel member of the carbonic anhydrase family, and while little is known about its physiological function, 33 it is expressed in the kidney, indicating a potential role in blood pressure regulation. 34 To our knowledge, the cellular target for mefruside’s mode of action has not been reported. 35
The mechanism of action for vadimezan/ASA404 is still not completely understood but has been proposed to act through the mitochondrial and endoplasmic reticulum protein STING. SEC-TID profiling revealed FPPS as a hit, and subsequent validation confirmed that ASA404 is in fact an allosteric inhibitor. Several reports in the literature are consistent with a model that the inhibition of FPPS, using known FPPS inhibitors such as zoledronic acid, brings about an increase in the phosphoantigen IPP. 36 FPPS normally catalyzes the production of (2E,6E)-farnesyl diphosphate using isopentenyl diphosphate and dimethylallyl diphosphate or geranyl diphosphate as substrates. Increases in IPP result in the activation of human Vγ9/Vδ2 T cells, which become more effective in the killing of tumor cells. ASA404 has also been shown to be effective both prophylactically and therapeutically against influenza. 37 Interestingly, IPP-activated Vγ9/Vδ2 T cells are also protective against influenza virus infection. 38 Therefore, it remains to be determined if ASA404 does indeed interact with FPPS in cells and whether modulation of FPPS activity contributes to its observed antitumor activity in mice. We confirmed that ASA404 interacts with murine FPPS (data not shown).
In summary, we believe SEC-TID is poised to make significant contributions to chemical genetics and drug discovery. At Novartis, we have observed that hits revealed from SEC-TID screening with small molecules with unknown molecular mechanism of action can quickly be triaged through integration of structure-activity relationships (SAR) and gene expression analysis. But ultimately, the success of this approach for small-molecule target identification will depend on having a much larger protein library representing broader coverage of the human proteome. We are tackling this challenge by emphasizing the production of domains from proteins predicted to interact with small molecules. But even when the actual efficacy targets are not present in the protein library, a protein domain-based analysis of small-molecule interacting proteins may lead to the efficacy target. For example, if Tnks1 and Tnks2 were not in our protein screening library when we screened for XAV939 interacting proteins, an analysis of the hits reveals that PARP2 has a PARP domain. Through subsequent protein production of PARP domains from proteins not present in the library and testing for interaction with XAV939, such an approach would have eventually revealed that the PARP domains of both Tnks1 and Tnks2 are high-affinity interactors for XAV939. To go beyond the limitations of using just purified proteins, further exploration into the use of protein complexes, protein fractions, or complex mixtures obtained from small-molecule responsive cells or tissue is also likely to aid in target identification. For example, if a cellular fraction contains a component that interacts with a small molecule in SEC-TID that demonstrates SAR, further fractionation and identification of interacting macromolecules by either mass spectrometry or next-generation sequencing technologies could lead to novel protein or nucleic acid targets. Last, SEC-TID may have greater applicability in drug discovery in early hit selection and preclinical safety profiling because the identification of molecules that interact with fewer targets, or target classes, could be prioritized for the value of simplifying target validation or for increasing the probability of lower compound-induced toxicity.
Footnotes
Acknowledgements
We thank Jason Thomas for assistance in bromodomain binding analysis for +JQ1. We are grateful to Sandra Nunes for providing human DHODH. We would especially like to thank several members (Nicola Burgess-Brown, Opher Gileadi, Susanne Graslund, Yanjun Li, and Katarzyna Kupinska) of the Structural Genomics Consortium for providing plasmids. Without their support, much of the work reported in this article would not have been possible. We would like to thank Travis Stams and Markus Schirle for reviewing the manuscript. We are also thankful to Stephen J. Haggarty at Harvard University for initial brainstorming discussions that focused on small-molecule selection and potential for the technology.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Novartis Institutes for Biomedical Research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.