
Abstract
During viral infection of human cells, host kinases mediate signaling activities that are used by all viruses for replication; therefore, targeting of host kinases is of broad therapeutic interest. Here, host kinases were globally screened during human influenza virus (H1N1) infection to determine the time-dependent effects of virus infection and replication on kinase function. Desthiobiotin-labeled analogs of adenosine triphosphate and adenosine diphosphate were used to probe and covalently label host kinases in infected cell lysates, and probe affinity was determined. Using infected human A549 cells, we screened for time-dependent signal changes and identified host kinases whose probe affinities differed significantly when compared to uninfected cells. Our screen identified 10 novel host kinases that have not been previously shown to be involved with influenza virus replication, and we validated the functional importance of these novel kinases during infection using targeted small interfering RNAs (siRNAs). The effects of kinase-targeted siRNA knockdowns on replicating virus levels were measured by quantitative reverse-transcription PCR and cytoprotection assays. We identified several novel host kinases that, when knocked down, enhanced or reduced the viral load in cell culture. This preliminary work represents the first screen of the changing host kinome in influenza virus–infected human cells.
Introduction
Kinases represent the single largest mammalian enzyme family with more than 500 kinase members in the human proteome. 1 Regulation of host processes is in large part controlled by the cellular kinases and other adenosine triphosphate (ATP)-hydrolyzing enzymes. Posttranslationally, these kinases modify the activity or function of target proteins (by phosphorylation of their substrates) and play a central role in regulating processes such as cell division, inflammation, angiogenesis, and metabolism by modulating signal-transduction pathways. 2
During viral infection of human cells, protein and lipid kinases play major roles in virus propagation and represent broad targets for the interruption of multiple virus life cycles. Following infection, the goals of the host cell are the immediate detection of virus invasion and the mounting of internal immune responses to blunt virus replication and harden neighboring cells against subsequent infection. In contrast, the goals of the virus are to combat the cellular response to infection and to highjack cellular processes and redirect them toward virus replication. This contention between host and virus is arbitrated by the host-signaling pathways that control cellular processes and whose regulation is altered in the presence of the virus and its gene products. These processes include developmental and apoptotic pathways, cell proliferation, cytoskeleton and membrane dynamics, and in particular cellular immune and antiviral responses. 3
Despite intense efforts, the physiological regulation and interplay of the majority of host protein kinases during virus infection and replication remain a largely unknown and yet vitally important process for mediation of viral disease. Because these kinases may be used by multiple viruses, effective targeting of host-signaling cascades may lead to broadly active therapeutics. 4 Another major benefit of this strategy is that viruses do not genetically encode the targeted host kinases and are expected to be less likely to develop the resistance associated with therapeutic inhibition of viral targets. 3 Last, new host-target identification will lead to new assays and the generation of novel therapeutic compounds.
Here, we globally screened host kinases in influenza-infected and uninfected A549 (ATCC CCL-185) cells and compared the profile signal changes within a pool of 268 human cellular adenine nucleotide–binding proteins (listed and classified in
A549 cells were infected at a high multiplicity of infection (MOI = 10) with seasonal influenza virus strain A/Brisbane/59/2007. The influenza virus model has been previously used to identify a limited number of novel targets for antiviral drugs that inhibit viral replication by disrupting signaling pathways. 7 This study was accomplished by using a recently established method for labeling and identifying active adenine nucleotide–binding proteins. 8 Using time-dependent sampling, we identified kinases within the human kinome whose ATP- and adenosine diphosphate (ADP)-analog probe-binding affinity was increased or decreased by influenza virus infection and replication. We hypothesized that observed changes in the kinase probe affinity suggest novel host molecular targets that affect viral propagation, may have potential as new targets for antiviral therapeutics, and provide a valuable understanding of how viral infection deregulates kinases in the host cell. We then validated the impact of the affinity changes of these kinases on viral replication by inhibiting their individual expression using targeted siRNAs and determining the effect of target knockdown on viral replication. We identified a series of kinases whose knockdown either enhanced [measured 24 h post infection (p.i.)], or reduced (measured 72 h p.i.) the number of total viral particles released into the cell supernatants. This study has the potential to broaden the list of human host kinases that affect influenza replication, alter cellular metabolism and signaling, bias the immune response toward nonproductive activities (e.g., cytokine storm), and may ultimately be validated as therapeutic targets.
Materials and Methods
A549 Cell Culture and Infection with Seasonal Influenza Viruses
A549 cells were grown to ~70–90% confluence in tissue culture flasks, and then infected with A/Brisbane/59/2007 (H1N1). Cells were infected at a high MOI of 10 in the absence of trypsin. Approximately 0.5 mL of pelleted A549 cells (eight T225 flasks) was needed for analysis at each time point. Cells were maintained and plated in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and 1% Pen/Strep/Glutamine (PSG; Invitrogen). Twenty-four hours after plating, the flasks were infected or mock-infected with influenza. The growth media were removed, and cells were washed 3× with Dulbecco’s Phosphate-Buffered Saline (DPBS). A 1:50 dilution of the virus in DMEM–PSG was added to flasks, and they were returned to 37°C/5% CO2 for 1 h. After this time, the flasks were removed, the infection media were replaced with DMEM–PSG–FBS, and the flasks were returned to the incubator. Cells were incubated in the presence of virus and harvested at 1, 4, 8, 12, or 24 h. As a negative control, uninfected cells were subjected to the same procedures to establish a basal level of kinase activities. Samples collected at the noted hours p.i. were labeled with ATP and ADP probes, and then digested with trypsin prior to analysis (detailed in this article and in Patricelli et al. 8 ).
General Procedures for Kinome Sample Preparation
The cells were removed, washed once in media to inactivate the trypsin, washed several times in DPBS, and centrifuged at low speed (1000× g). The cell pellet was resuspended in 1× lysis buffer [25 mM Tris pH 7.6, 150 mM NaCl, 1% CHAPS, 1% Tergitol, 1 mL Phosphatase Inhibitor Cocktail II (EMD/Calbiochem)], and homogenized 15 times with a Dounce homogenizer on ice, followed by sonication twice for 10 s to complete cell lysis. The sample was centrifuged at 12,000 rpm for 30 min in a microfuge, and then centrifuged at 33,000 rpm for 30 min in an ultracentrifuge. Finally, the sample was passed through a 0.2 µM syringe filter before size-exclusion chromatography. Size-exclusion chromatography was performed to remove endogenous nucleotides and further purify the samples. A BioRad 10DG column was used. The column was equilibrated with buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% Triton-X-100) and then drained, followed by the addition of up to 2 mL of sample volume. Fractions were taken in 0.5 mL aliquots and assayed by BCA assay for protein concentration. Aliquots containing high protein concentrations (5–15 mg/mL) were pooled for the labeling reaction.
Sample Labeling
All methods are outlined in Patricelli et al.8,9 ATP and ADP acyl-phosphate probes were synthesized as described previously except that desthiobiotin was substituted for biotin in all cases.
Sample Preparation for Liquid Chromatography–Tandem Mass Spectrometry, and Analysis
Samples were prepared and analyzed on an ion-trap mass spectrometer coupled with Agilent 1100 series micro-HPLC (high-performance liquid chromatography) systems with autosamplers.
Peptide Sequence Alignments and Generation of Sequence Logos
Peptide sequences were performed using protein-kinase sequences obtained from http://www.kinase.com/ATPome and aligned using ClustalX. Sequence logos were generated with the Delila suite of programs.
Mass Spectrometry (MS) Data Analysis
All MS data were analyzed using custom software programs. Briefly, runs to be analyzed were aligned and normalized in both time and intensity dimensions using MS data obtained from each sample. Because the probes label a large number of kinases and other non-kinase ATPase proteins, signals were normalized based on the averaged signal ratios of major parent ions throughout the run. Signal normalization factors were typically less than 30%. For ion selection, a “reference spectrum” concept was used. Aggregate reference spectra were generated by extracting and averaging the highest scoring spectra (up to 20) for each peptide to be analyzed based on historical data. These reference spectra allow for peak assignment based on both the presence and intensity of expected ions. Typically up to four ions were selected for signal extraction and quantization based on their presence, intensity, and correlation to the reference spectrum. The resulting chromatographic peaks from each run were then integrated, and the integrated peak areas were used to determine percentage inhibition values relative to control runs.
Optimization of siRNA Transfection into A549 Cells
siRNAs were individually ordered from Qiagen (Flexiplate format; Qiagen) and were reconstituted to 10µM in sterile nuclease-free water (Fisher Scientific) per Qiagen directions. Aliquots were further diluted in sterile nuclease-free water to 20 nM and stored at −20°C. Transfection with essential gene siRNAs was optimized to conditions that result in >90% cell death after 72 h. Using the optimized procedure, 5 µL siRNA was spotted directly into the bottom of a 96-well plate, and 25 µL of a mastermix containing Hiperfect (Qiagen) and growth medium (DMEM + PSG + 0.1% bovine serum albumin) was added to the siRNA. Complexes were allowed to form for 5–10 minutes at room temperature before the addition of A549 cells in corresponding media. Controls used to measure the efficiency of the transfections included untargeted siRNAs and siRNAs targeting essential host-survival genes (Death Control; Qiagen catalog no. SI04381048). Cell viability was measured using CellTiter-Glo (Promega).
Influenza Virus Detection Assays
In the profiling experiments, the kinase probe-affinity signals measured in triplicate control samples did not vary greater than +/− 1.5-fold. Therefore, kinase activities that varied by greater than 1.5-fold in duplicate infection experiments were selected for validation by targeted siRNA knockdown. siRNA target validation was done by transfecting a proprietary mixture of 3–4 siRNAs (Qiagen) for each target in a 96-well format followed by viral infection and analysis of viral replication. As a control for cell viability, one plate of siRNA-transfected, uninfected cells was tested in parallel. Following a 72 h siRNA transfection, A549 cells were infected with influenza strain A/Brisbane/59/2007 at an MOI of 0.005 (using the procedures described in Noah et al. 10 ) and incubated at standard conditions. Media from the infected wells were sampled at time points of 24, 48, and 72 h, and the numbers of viral particles in the supernatants were determined by quantitative reverse-transcription PCR (qRT-PCR). 11 Cytoprotection assays were performed as described in Noah et al. 10
qRT-PCR Assays
One microliter of infected cell culture supernatant was combined with 12.5 µL of 2× reverse-transcription reaction buffer (Invitrogen SuperScript® III One-Step RT-PCR System with Platinum®Taq, Cat. No. 12574-026) and heated to 95°C for 2 min. The mixture was then combined with the primer set targeted to the M gene of the influenza virus (25 pmoles each: 5′-AGATGAGTCTTCTAACCGAGGTCG-3′ and 5′-TCGA-GATCGGTGTTCTTTCC-3′) and probe (6FAM-TCAGGC-CCCCTCAAAGCCGA-MGBNFQ), with enzyme mix, and brought to a final volume of 25 µL with nuclease-free water. qRT-PCR was performed on a Mastercycler EP Realplex real-time PCR system (Eppendorf). qRT-PCR data shown are averaged from three independent experiments.
Kinase-Pathway Mapping
Kinase pathways were mapped using the open-source platform Cytoscape 2.8. 12 Central kinases and their nearest interacting neighbors were mapped using the MiMi plugin. 13
Results
Kinase Profiling of A549 Cells Infected with the Human H1N1 Influenza Virus Reveals Modest Changes in Global Host-Kinase Probe Affinity during the First 24 H Post Infection
We examined the signal profile of a pool of 268 human cellular adenine nucleotide–binding proteins (listed and classified in
A549 cells were infected at a MOI of 10 with seasonal influenza virus strain A/Brisbane/59/2007. This high MOI was used to ensure that the virus infected the majority of cultured cells simultaneously, therefore allowing us to maximize the measured global kinase probe-affinity changes and reduce assay noise resulting from an asynchronous infection. By probing for expression of the influenza M2 protein, we verified that, at the MOI used, ~90% of cells were infected and robustly producing viral proteins (data not shown).
At various time points (1, 4, 8, 12, and 24 h p.i.), we harvested the infected and uninfected control cells and prepared lysates for profiling. Acyl phosphate–containing nucleotides were used as probes. ATP and ADP probes were both used to maximize coverage in a diverse target space. Cell lysates were depleted of endogenous ATP, probed with the activated nucleotide probe, and treated with trypsin to produce desthiobiotinylated peptide fragments from the labeled kinases. These peptides were affinity captured, identified, and quantified using an MS-based analysis platform. The quantization of a subset of peptides from cellular kinases was established by comparing the results of uninfected control cells with those of infected cells, with the rationale that kinases with altered probe affinities during infection might reflect virus-induced changes in kinase expression, activation state, or activation-independent changes in the ATP-binding site. A time-dependent profile of differential probe affinities was established for 29 human kinases whose quantitative-labeling was consistently (observed in at least two independent experiments) up- or downregulated during the first 24 h p.i. Due to limitations in the methodology used, the very early changes in probe affinities that take place before the earliest measured time point (<1 h) were not determined.
Human H1N1 Influenza Virus Infection Alters the Activities of Multiple Unreported Host Kinases
Three independent kinome screens were performed. Affinity changes ranged from 1.5 fold to >fivefold. Signal changes <1.5 were not considered significant. Kinases whose probe affinity significantly changed (i.e., a 1.5-fold or greater increase or decrease at any single time point) in duplicate, independent profiling experiments were selected for additional investigation. Figure 1 shows the results from a representative kinase-profiling experiment and lists the kinases that showed altered probe affinities during influenza virus infection. The relative changes in kinase signal at each specific time point (1–24 h p.i.) at which cellular lysates were prepared and probed with the ATP or ADP analogs are shown. In each case, the probe yielding the greatest consistent affinity change is indicated. The table is divided into two different categories (previously implicated host kinases and novel host kinases). The novel host kinases are further categorized based on subsequent targeted siRNA knockdown results for each listed kinase (discussed below).
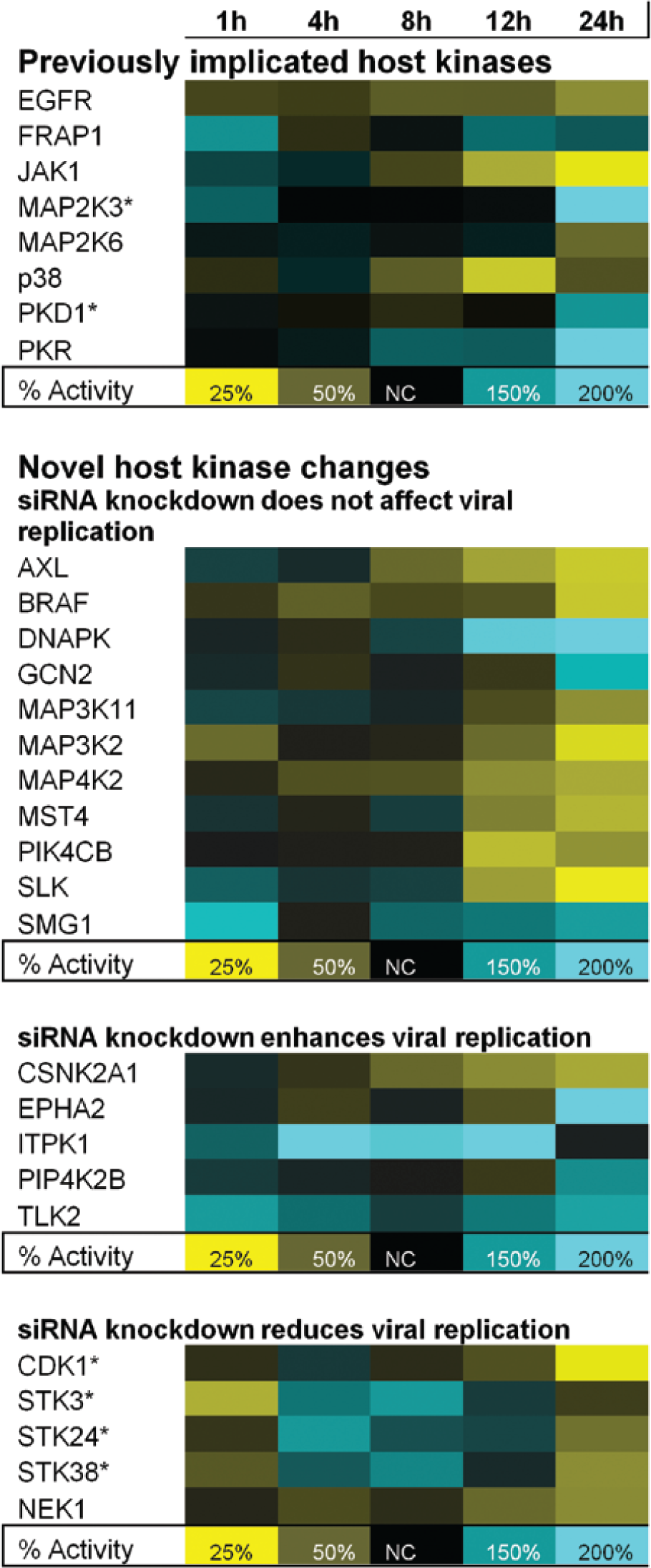
Host kinase activity changes during H1N1 infection. Blue indicates increased activity, yellow indicates decreased activity, and black indicates no change (NC) in activity. Percentage of activity is compared to the control, uninfected cells. NC indicates that the measured kinase activity in infected cells was 100% of that observed in the infected control cells. An asterisk (*) indicates that the adenosine diphosphate probe was detected and used to measure changes in host kinase activity.
Several of the listed kinases ( Fig. 1 , top) have been shown by other reports (reviewed in Meliopoulos et al. 14 ) to be involved in the influenza virus life cycle, and they were not further investigated in this study. These previously implicated kinases were also identified in this study and provide convincing validation that the screening method can accurately identify changes in signal for kinases that are involved in viral infection.
Targeted siRNA Knockdown of 10 Host Kinases Affects Influenza Replication
Several kinases identified in the screens have not been previously shown to be affected by virus infection. Additional kinases that showed consistent activity changes during influenza virus infection are also listed in Figure 1 (middle and bottom). These kinases were further investigated to assess their particular importance to the influenza virus life cycle and as potential therapeutic targets. This was done using sequence-specific siRNAs to reduce the translational expression of individual kinases before and during viral infection. Targeted siRNAs were reverse-transfected into A549 cells for 72 h, followed by infection at a low MOI (0.005) by H1N1 influenza virus. Because a simultaneous infection was not required at this step, and to measure the effects of siRNA targeting on early, middle, and late replication stages, a low MOI was used to allow for multiple rounds of virus infection and replication. At 24, 48, and 72 h p.i., qRT-PCR was used to measure the number of viral particles released into the supernatant of the transfected, influenza-infected cells. A sequence-scrambled, nontargeting siRNA and siRNAs targeted to essential genes were included as experimental controls for both experimental perturbations and reverse-transfection efficiency, respectively. Three independent experiments were performed for each knockdown. At 72 h p.i., virus titers (genomic copy number) in the supernatants of transfected and infected cells were analyzed by qRT-PCR and compared to mock-transfected and uninfected controls or mock-transfected and infected controls.
The results of the validation procedure allowed us to classify the novel kinases into three groups: those whose targeted siRNA knockdown had no effect on total viral particles, those whose knockdown reduced total viral particles, and those whose knockdown increased total viral particles. Figure 1 (center) shows novel human kinases whose probe affinity profiles are consistently altered during the first 24 h of influenza virus infection, but whose targeted siRNA knockdown has no effect on the course of viral infection. These were not further investigated. Figure 1 (bottom) also shows kinases whose siRNA-mediated knockdown resulted in either an increased viral particle number or a decreased viral particle number.
Validation of the effects of siRNA knockdown of individual host kinases on viral replication during influenza infection is shown in Figures 2 , 3 , and 4 . Figure 2A shows the relative numbers of viral particles detected in the supernatants of cells transfected with siRNAs targeted to individual host kinases, measured 24 h p.i. Specific knockdown of each of the indicated kinases resulted in a higher viral copy number at this time point, compared to the control experiment that was reverse-transfected with a scrambled, nontargeted siRNA. These kinases are CSNK2A1, EPHA2, ITPK1, PIP4K2B, and TLK2. Figure 2B shows time-dependent growth curves during targeted knockdown for each of the kinases investigated. A significant siRNA-mediated increase in viral particles (10-fold) was observed only at the 24 h time point, and viral particle values at 48 and 72 h showed no significant difference between the targeted and nontargeted siRNAs. Figure 2C shows the corresponding viability of targeted siRNA-transfected cells at 72 h p.i., compared to cells transfected with a scrambled, nontargeted siRNA as a control. Because A549 cells do not show significant cytopathic effect in response to seasonal human influenza virus infection, the cytoprotection assays were used to reveal any cellular toxicity that resulted from siRNA-mediated knockdown of the targeted kinases. The viability of infected and uninfected cells is compared, and this comparison shows that none of the targeted siRNAs were toxic to uninfected cells (viability of 90–120% of control) at 72 h post transfection. For infected cells, only siRNAs targeted to PIP4K2B caused a significant decrease in cell viability (45% of control). Mock transfection without siRNAs had no effect on cell viability (data not shown).
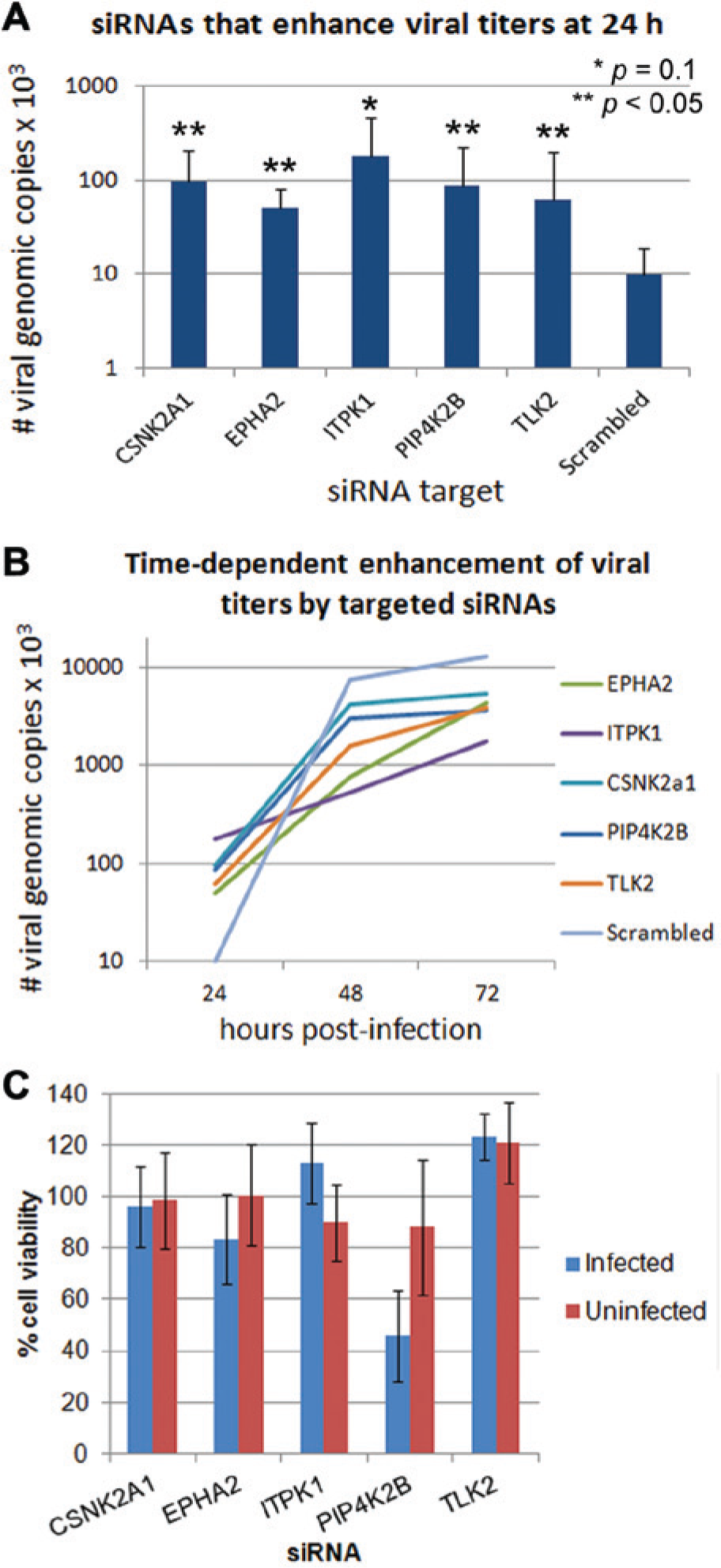
Small interfering RNA (siRNA) knockdown of kinases resulted in increased viral replication. (
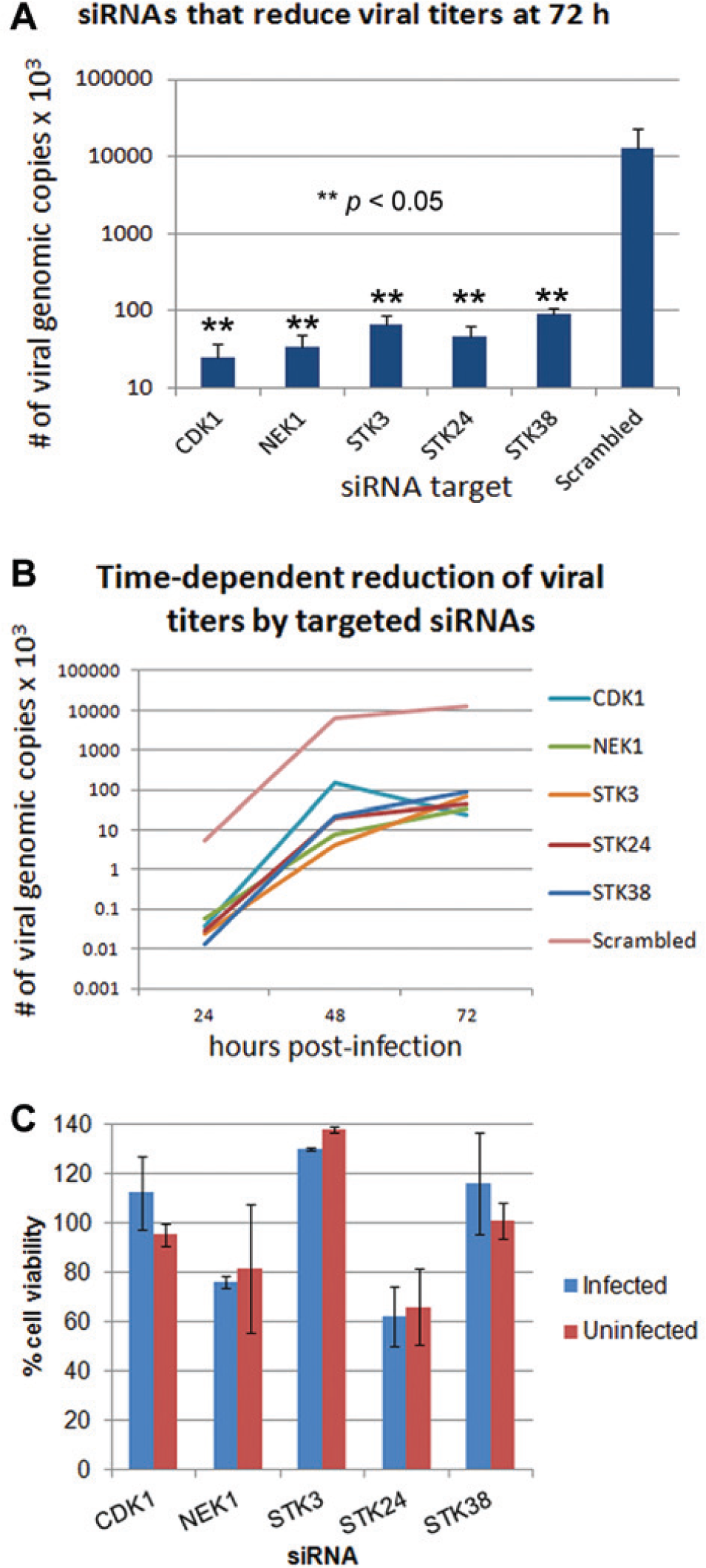
Small interfering RNA (siRNA) knockdown of kinases resulted in decreased viral replication. (

Verification of siRNA-mediated knockdown of protein kinase R (PKR) expression by Western blotting at 72 and 144 h post transfection. “Tranfect. reagent” indicates transfection reagent. PKR indicates the protein kinase activated in response to double-stranded RNA. “Scrambled siRNA” indicates a nontargeting siRNA used as a control.
Figure 3A shows each of the kinases whose specific knockdown resulted in a significant decrease (~100-fold) in viral particles released (compared to a scrambled siRNA) when measured 72 h p.i. These kinases are STK38, STK24, NEK1, CDK1, and STK3. Figure 3B shows the time-dependent growth curves during targeted knockdown for each of these kinases. The siRNA-mediated decrease in viral particles was observed at all measured time points, including at 48 and 72 h p.i., when compared to the control experiments. Figure 3C shows the corresponding viability of targeted siRNA-transfected cells at 72 h post transfection, compared to cells transfected with a scrambled, nontargeted siRNA as a control. The viability of infected and uninfected cells is compared, and it shows that none of the targeted siRNAs were toxic to uninfected cells (viability of 80–140% of control) except for STK24, which decreased uninfected cell viability by ~40%. For infected cells, these results were recapitulated.
siRNA-Mediated Knockdown of Targeted-Kinase Expression and Probe Affinity Was Verified by Western Blot and Global Profiling
To verify that the siRNA reverse-transfection techniques were resulting in efficient knockdown of gene expression, siRNAs to essential genes (proprietary to Qiagen) were included as controls in each experiment. siRNA transfection targeted to these genes routinely resulted in a 99% decrease in cell viability for reverse-transfected cells. We also verified that siRNA transfection resulted in a reduction of the expression of a targeted kinase using Western blotting. We chose PKR as a control kinase known to be upregulated by influenza virus infection. Measurement of PKR expression levels at 72 and 144 h post transfection in infected cells is shown in Figure 4 . It demonstrates that in each PKR-targeted siRNA transfected lane, we were able to completely reduce the expression of PKR up to 144 h post siRNA transfection, supporting that the methodology was successful in targeting specific proteins and reducing gene expression throughout the time course of the experiments. This effort also confirmed that neither the scrambled, nontargeting control siRNA nor the transfection reagent alone had an effect on PKR expression.
To confirm that transfection of targeted siRNAs also resulted in a reduction of probe affinity of the targeted kinase (as a result of decreased target expression) throughout the time course of the experiments, we measured the global affinity profile of the targeted kinases at two different time points, post tranfection in uninfected cells, using the primary global-profiling method. Individual, targeted siRNAs were reverse-transfected into uninfected cells, and the global activities of cellular kinases were measured 72 and 144 h later using ATP-analog probes. At these time points, cellular lysates were prepared, and the relative activities of kinases were measured. The results confirmed the efficient target knockdown by siRNA transfections. Table 1 shows that, in independent experiments at 72 and 144 h post siRNA transfection, significant reductions in probe affinity are observed for each target kinase. In these experiments, nine of the 10 kinases investigated in Figures 2 and 3 were detected; CNSK2A1 was not detected. The probe affinity ranged from 14% to 33% (depending on the target) at 72 h post transfection. The siRNA-mediated reductions of kinase affinities were greatest at 72 h post transfection; however, significant reductions were confirmed for a subset of measured kinases at 144 h post transfection. Our overall conclusion was that we were able to significantly reduce target ATP-binding affinity by siRNA transfection due to a decrease in target protein expression.
Percentage Kinase Probe Affinity at 72 and 144 H post siRNA Transfection.*
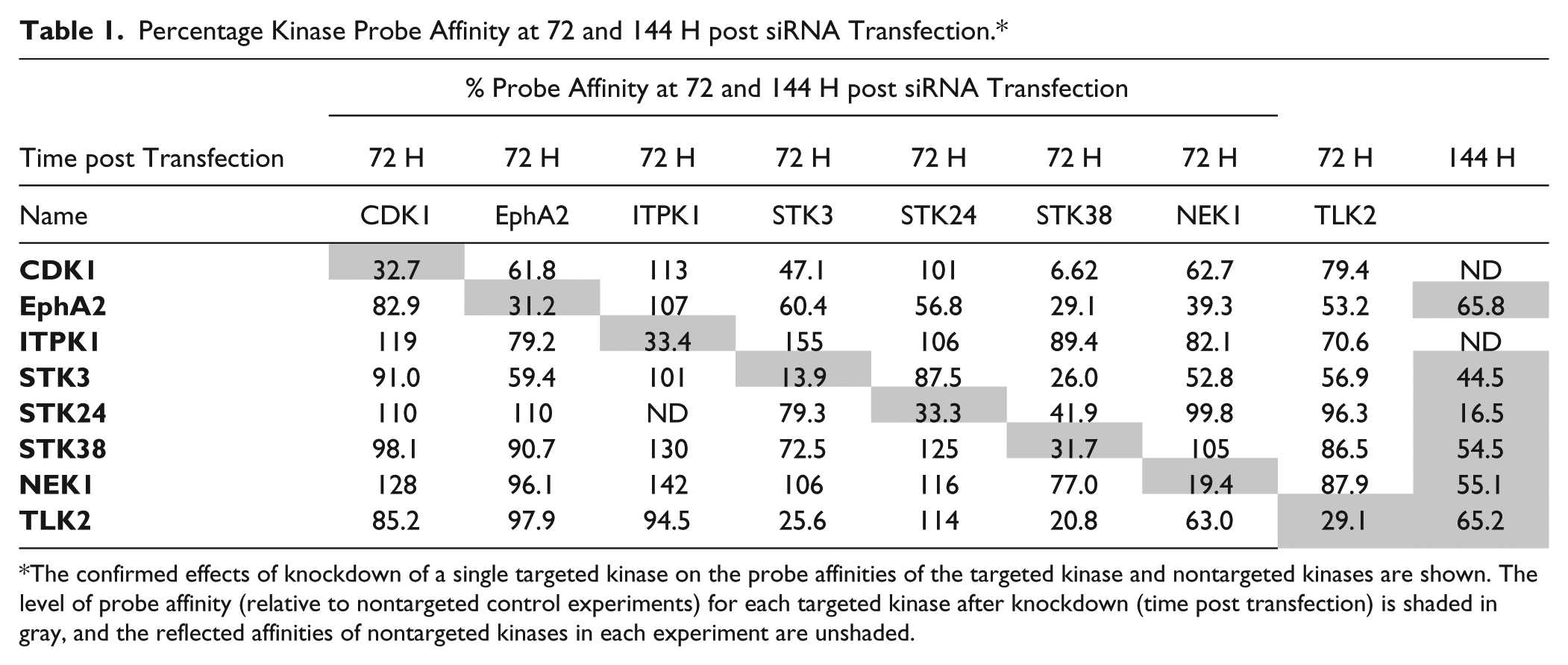
The confirmed effects of knockdown of a single targeted kinase on the probe affinities of the targeted kinase and nontargeted kinases are shown. The level of probe affinity (relative to nontargeted control experiments) for each targeted kinase after knockdown (time post transfection) is shaded in gray, and the reflected affinities of nontargeted kinases in each experiment are unshaded.
Table 1 also shows the more global effects of single-kinase knockdown on the measured activities of the other kinases in this set. Equating probe affinity with the siRNA transfection in this manner also allowed us to measure the off-target effects of targeted siRNAs. That is, we were able to quantify the effects of siRNA transfection on nontargeted kinases within our subset of interest in the context of the affinity-probing experiment. In most cases, the targeted knockdown resulted in specific reductions in target probe affinity without significant effects on the other kinases. But for two of the targeted kinases (STK38 and NEK1), targeted siRNA knockdown resulted in multiple and dramatic decreases in the affinities of nontargeted kinases within the examined profile. For NEK1, this result may explain the reduction in uninfected and infected cell viability for cells that were transfected with NEK1-targeted siRNAs ( Fig. 3B ). Although targeting STK38 also resulted in significant decreases in the signal profile of multiple other kinases, cell viability was not affected ( Fig. 3B ).
Discussion
Multiple siRNA screens 14 have used varied methods and reporters to identify host genes involved in influenza replication. Each screen generated a list of potentially important host genes, but these lists did not greatly overlap. This suggests that factors such as virus strain, cell type, reporter construct and endpoint, and the source and efficiency of siRNAs contribute to significant experimental variation. In support of this, studies by Karlas et al. 5 and Konig et al., 6 both of which used A549 cells and the A/WSN/33 virus strain, show the highest degree of overlap. Given the observed variability, a more global and physiologically relevant approach to screening is required. This study was designed to address this by comprehensively surveying approximately ~300 human kinases to identify those that participate in or affect the influenza virus life cycle. Because kinases have already been shown to be critical during influenza infection, our goal was to observe the global changes in host kinases probe affinity (which can be correlated with kinase activity). 15 In light of previous screens, another siRNA screen may not have yielded additional information, so our study was an attempt to complement the results of siRNA screening and demonstrate that such global approaches can be successful. We used an influenza infection model and probed for active kinases that (1) control or promote the cellular antiviral response, and (2) are active in the uninfected host cell but are crucial for viral infection and replication. Given that kinases generally are considered to be good druggable targets, 16 we used this approach to identify new molecular host targets that can be incorporated into biochemical screens for new drug discovery.
In this global kinase-screening experiment, a probe (
At various time points (1–24 h p.i.), cellular lysates were probed for kinase affinity. Lysates were purified, probed, and labeled, and then treated with proteases. Labeled peptide fragments were captured, identified, and quantified using a mass spectrometry–based analysis platform. The relative amounts of a subset of cellular kinases were determined by comparing the signal from uninfected control cells with that of infected cell samples. A time-dependent signal profile was established for 27 human kinases whose probe affinities were consistently up- or downregulated during the first 24 h p.i. To validate these kinases as targets, we performed siRNA transfections targeted to the specific host-kinase mRNAs prior to and during virus infection. The effect of targeted siRNA-mediated knockdown on viral load was measured by qRT-PCR and cytoprotection assays. We identified 10 kinase targets that, when knocked down, enhanced or reduced the total number of viral particles released in cell culture for H1N1 influenza.
Five host kinases, when knocked down, caused significant increases in the number of total viral particles in the supernatant only within the first 24 h of a multiple-replication-cycle experiment. Of these five, CSNK2A1 is known to phosphorylate various viral proteins, and also functions in cell proliferation, differentiation, and apoptosis. 17 EPHA2 mediates cellular developmental events and has been implicated as an altered region in human cancers. 18 ITPK1 is known to modify TNFα-induced apoptosis by interfering with the activation of TNF/RSF1A, and binds to CSN1. 19 PIP4K2B catalyzes the phosphorylation of phosphatidylinositol-5-phosphate. Additionally, the encoded protein interacts with p55 TNF receptor, and plays a role in cell proliferation. 20 Lastly, TLK2 is involved in chromatin assembly. 21 We did not further investigate the individual mechanisms by which these kinases may act to promote early increases in virus particle numbers.
Five host kinases, when knocked down, caused significant decreases in the number of total viral particles when measured after 72 h of a multiple-replication-cycle experiment. These kinases are more relevant as putative targets for antiviral intervention. CDK1 is a key serine–threonine kinase that controls cell cycle regulation and progression by binding cyclins. It is known to interact with multiple host-essential proteins. 22 STK3 is a pro-apoptotic kinase that functions as a cellular growth suppressor. 23 STK24 functions upstream of the mitogen-activated protein kinase (MAPK) cascade. 24 STK38 is a negative regulator of the MAP3K1/2 pathways, which contribute to cytokinesis, cell proliferation, and apoptosis. It is stimulated by STK24.25,26 NEK1 is associated with arrest of cellular development during DNA damage and repair. NEK(−) cells do not arrest in the G(2)/M-phase if DNA damage occurs. 27
From the existing literature, direct mechanistic links between these host kinases and influenza replication are not clear. Processes such as cytokine generation, cellular development, cytokinesis, and apoptosis are certainly indirectly related because these modulating processes are known to support or counter virus growth. To suggest additional mechanistic insight and investigate any published associations of the five kinases that reduced total viral particles, we used Cytoscape, 12 an open-source software tool for the analysis of bimolecular interaction networks. We coupled Cytoscape with databases of protein–protein interactions through the MiMI plugin. 13 MiMI integrates data from multiple well-known protein-interaction databases using an intelligent deep-merging approach. This facilitated access to the molecular interaction data assembled in MiMI from the Cytoscape user interface, and it allowed us to readily explore and analyze interaction data from MiMI by using a nearest-neighbor parameter. The nearest-neighbor parameter displays the direct protein interactions using published literature information associated with those interactions from multiple well-known protein-interaction databases. 13 Figure 5 shows the nearest-neighbor mapping of STK3, STK24, STK38, and NEK1, which are four of the kinases that, when targeted by siRNAs, result in reduced influenza particle numbers in cell culture. The fifth member of this group, CDK1, was omitted due to the extensive interactions reported for this kinase, which made mapping impractical. Our mapping shows a tight grouping of the four kinases and a clear nearest-neighbor relationship between them. Mapping such as this is useful for suggesting additional host factors (based on evidence in literature) that are associated and may also be antiviral targets or participate in kinase pathways that are critical for influenza virus replication. In support of this hypothesis, we used a siRNA to HN4FA, which was a transcription factor central in our mapping analysis, to test the effects of its knockdown on viral replication. This host factor has not been previously shown to be linked to influenza infection. 28 In our experiments, targeting this factor with siRNAs was highly toxic to A549 cells (results not shown), thereby highlighting the necessity of target validation. Additional targets in the map are currently being tested.
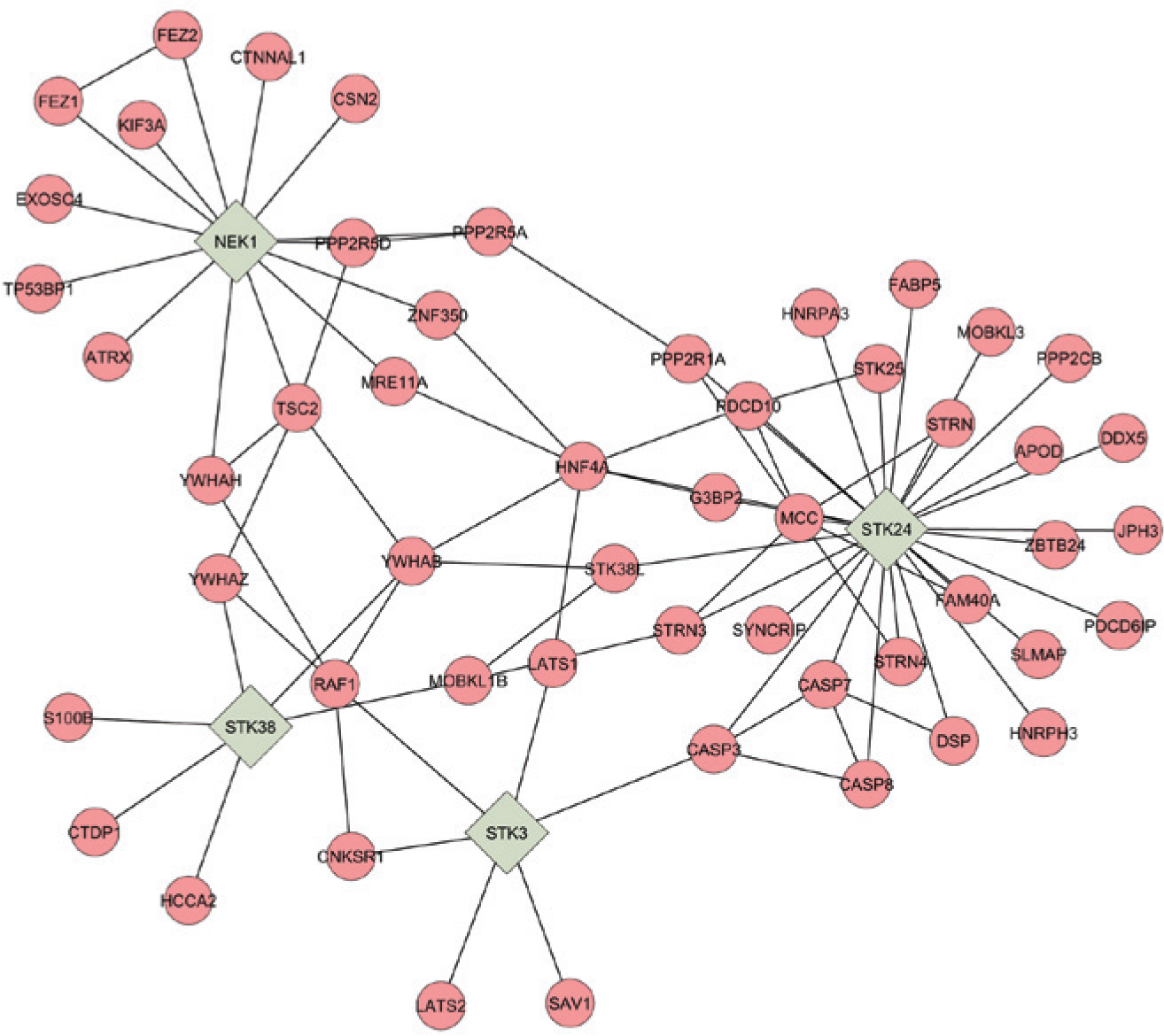
A nearest-neighbor network of STK3, STK24, STK38, and NEK1. The small interfering RNA (siRNA)-mediated knockdown of these human host kinases during viral infection in A549 cells resulted in decreased influenza replication. Molecular interaction data were assembled using the MiMI plugin 13 to the Cytoscape user interface 12 using a nearest-neighbor parameter based on sorted literature reports.
The global kinase-screening methods described in this article are an innovative approach to identifying novel and druggable infectious-disease targets. Recently, novel siRNA screens for profiling genetic regulation and transcriptional activity have been developed that have allowed for the generation of tremendous amounts of biologically important information. 14 However, genetic screens do not reveal the importance of posttranslational regulation such as modifications, interactions with other proteins, and subcellular localization. Similarly, proteomic approaches to profiling protein levels and expression do not strongly link the functional aspect and activity of the putative targets, and hence do not link profiling data to drug-target identification and subsequent drug discovery. Here, we have attempted to generate a map of kinases whose activities are linked to viral replication. This effort suggested that a number of the identified kinases may participate in nearest-neighbor relationships and therefore represent a condensed host pathway that could be successfully targeted at multiple points to maximize kinase inhibitor-based antiviral strategies. Given the highly deregulated cytokine levels evident in avian influenza infections, 29 global screens of host kinases in infected systems may reveal key points of intervention and mitigation of the cytokine storm. This preliminary work represents the first screening, assembly, and evaluation of the human kinome of influenza virus–infected cells and is the basis for further investigations of influenza infection using kinase inhibitors and additional virus strains (e.g., highly pathogenic avian influenza). Finally, this work could be readily adapted to validate a new approach for exploring a wide variety of host–virus interactions with a broader range of viruses. Repeated studies of this nature with a representative strain from multiple virus families, all in human cell lines, would provide a global-activity map of relevant pathogen–kinase interactions that would complement existing genomic and proteomic maps.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The project described was supported by Grant No. U54HG005034 from the National Human Genome Research Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Human Genome Research Institute or the National Institutes of Health. This study was also supported in part by a grant to UAB from the Howard Hughes Medical Institute through the Med-into-Grad Initiative.