
Abstract
Plants represent a tremendous structural diversity of natural compounds that bind to many different human disease targets and are potentially useful as starting points for medicinal chemistry programs. This resource is, however, still underexploited due to technical difficulties with the identification of minute quantities of active ingredients in complex mixtures of structurally diverse compounds upon raw phytomass extraction. In this work, we describe the successful identification of a novel class of potent RAR-related orphan receptor alpha (RORα or nuclear receptor NR1F1) agonists from a library of 12,000 plant extract fractions by using an optimized, robust high-throughput cell-free screening method, as well as an innovative hit compound identification procedure through further extract deconvolution and subsequent structural elucidation of the active natural compound(s). In particular, we demonstrate that neoruscogenin, a member of the steroidal sapogenin family, is a potent and high-affinity RORα agonist, as shown by its activity in RORα reporter assays and from its effect on RORα target gene expression in vitro and in vivo. Neoruscogenin represents a universal pharmacological tool for RORα research due to its specific selectivity profile versus other nuclear receptors, its excellent microsomal stability, good bioavailability, and significant peripheral exposure in mouse.
Introduction
New target validation has always been a challenge for the pharmaceutical industry with respect to the potential therapeutic value. Information obtained through genetic gain- and/or loss-of-function approaches is of course of great value, but it does not replace target validation through pharmacological stimulation or inhibition of a target with selective and potent chemical ligands. The identification of such ligands usually takes 18 to 24 months or even more in programs where medicinal chemistry starts from high-throughput screening (HTS)–derived hit compounds identified in chemical libraries. Since it is essential to provide high-quality proof-of-concept validations early in the drug discovery process, technologies that enable the identification of potent, selective, and bioavailable chemical probes are of crucial interest, even if the resulting compounds require further optimization for development into a pharmaceutical product.
It has been recognized that natural compounds bear unique drug-likeness properties due to their immense structural diversity and their inherent propensity to interact with biological targets. Numerous examples exist to show that plant biodiversity screening has led to the identification of novel drug candidates.1,2 Indeed, the property space of natural products does match very well with the property space of drugs on the market, 3 considering that molecules derived from natural products are at the origin of nearly half of all the small-molecule drugs that have reached the market from 2000 to 2010. 4 Despite the fact that methods compatible with high-throughput extract fractionation and target screening were initially described several years ago,5,6 technical difficulties associated with structural elucidation of minute quantities of specific compounds in a complex matrix of plant secondary metabolites have made natural product–based drug discovery procedures unpopular in the drug discovery industry.
Nuclear receptors (NRs) constitute a large family of ligand-activated transcription factors that work in concert with other nuclear proteins to regulate the expression of genes important for development and homeostasis. NRs bind directly to specific DNA response elements in the regulatory regions of their target genes and modulate the rate of transcription in a ligand-dependent manner. NRs have a conserved domain structure composed of a variable amino-terminal A/B domain, a central highly conserved DNA-binding domain (C domain) hinge region (domain D), and a carboxy terminal ligand-binding domain (LBD; E domain). Ligand binding induces a conformational change in the receptor. For an agonist ligand, these results in the dissociation of a corepressor(s) to be replaced by a coactivator protein, whereas for an inverse agonist, it induces a conformational change that results in a decreased affinity of the receptor for the coactivator protein(s) and in the inhibition of its constitutive activity. Consistent with their key biological roles, nuclear receptors serve as important drug targets, 7 and NR ligands represent 10% to 15% of the present pharmacopoeia. 8
RAR-related receptors (RORs), originally identified by virtue of their homology to retinoic acid receptors (RARs), have gained broad attention because of multiple phenotypes observed in the RORα-deficient staggerer mice, which carry a natural loss-of-function mutation in the RORα gene. The staggerer mutation and/or targeted RORα deletion both result in serious defects in cerebellum development, including dramatic loss of Purkinje cells and granule cells, leading to extensive neurodegeneration of cerebellum and severe ataxia.9,10 A variety of other systemic effects, such as circadian rhythm regulation, effects on glucose and lipid homeostasis, immune system regulation, and xenobiotic metabolism, were described in mice deficient for RORα in addition to its role in CNS development. However, the complexity of RORα biology is beyond the scope of this article, and interested readers are referred to an excellent recent review. 11
Previous studies examining the crystal structure of RORα identified cholesterol and cholesterol sulfate present in the ligand-binding pocket.12,13 Subsequently, 7-oxygenated sterols (7α-hydroxycholesterol, 7β-hydroxycholesterol, 7-ketocholesterol) and (24S)-hydroxycholesterol were identified as novel RORα ligands, with comparable nanomolar binding affinities,14,15 and recently the structures of synthetic RORα ligands, based on either the benzenesulfonamide or thiourea scaffolds, have been published.16–18 However, no potent and bioavailable ligands are available to date to address important questions about the complex biology of the RORα receptor in vivo. We demonstrate in this work that less conventional approaches, such as the identification of receptor ligands starting from a natural product library of plant extract fractions, are an efficient and effective method to identify potent and bioavailable pharmacological probes to be used for orphan target validation studies.
Materials and Methods
Recombinant Proteins Used in the Study
DNA sequences coding for the glutathione S-transferase (GST) fragment (AAA57101:1–238), GST-RORα fusion protein, or GST-BAP fusion protein were cloned into the pRSETA (Invitrogen, Carlsbad, CA, USA). The GST-RORα construct contains the RORα sequence that corresponds to NP_599023.1:139–523. The GST-BAP construct contains the sequence that corresponds to the bacterial alkaline phosphatase fragment NP_414917.2:26–471. The three open reading frames were subsequently subcloned into a proprietary protein expression vector, and the proteins were produced in Sf9 (High-Five) cells by GTP Technology (Labège, France).
The sequences of both the TIF2 (NCOA2) fragment (NP_006531.1:413–812) and BAP (NP_414917.2:26–471) were inserted in frame in the pRSETA (Invitrogen). Three LXXLL motifs present in the TIF2 (413–812) fragment were mutated one by one into LXXAA motifs by the classic site-directed mutagenesis method (QUICK Change II Kit; Stratagene, La Jolla, CA) to yield the His-TIF2mut-BAP sequence. Both His-TIF2-BAP and His-TIF2mut-BAP fusion proteins were produced in Escherichia coli, and the purification was achieved through the immobilized-metal affinity chromatography (IMAC) at GTP Technology.
Pull-Down Assay
The pull-down assay was performed in high-binding capacity streptavidin-coated plates (Pierce, Rockford, IL, USA), which were coated overnight with biotin-conjugated, affinity-purified anti-GST antibodies (Antibodies Online, Aachen, Germany). Final antibody dilution was empirically optimized for each lot. Plates were washed with TBS-Tween (20 mM Tris-HCl [pH 7.2], 150 mM NaCl, and Tween 0.05%), and the reaction was set up in the binding buffer (15 mM Tris-HCl [pH 8.0], 15 mM glycerol, 0.15 mM EDTA, 0.04% NP40, 22.5 mM KCl, 0.25 mg/mL fatty acid–free bovine serum albumin, 1 mM dithiothreitol, and 1 mM benzamidine), in a final volume of 200 µL. For each reaction, 0.12 µg GST-RORα protein and 0.10 µg TIF2-BAP protein were used. The antiprotease tablets from Roche (Basel, Switzerland) were added (1 tablet for each 50 mL of the buffer) to prevent protein degradation. All compounds were added as DMSO solutions. 7-Dehydrocholesterol (7DC, 10 µM) was used as a reference compound. Binding reaction was performed overnight at 4 °C. Binding plates were washed with TBS-Tween (20 mM Tris-HCl [pH 7.2], 150 mM NaCl, and Tween 0.05%). Lumi-Phos WB (Pierce; 50 µL per well) was added following the washing step, and plates were incubated for 30 min at 37 °C. The luminescent signal was read with the TECAN Ultra 384-well microplate reader (TECAN, Männedorf, Switzerland). The integration time was set to 500 ms.
To discard compounds that induce a nonspecific signal, hit compounds were subsequently tested in counterscreen assays. We have used assays that measured (1) a recruitment of a mutated TIF2-BAP protein on GST-RORα, (2) a recruitment of TIF2-BAP on a GST protein fragment that contains no ROR-LBD, and (3) the effect of the compound on a chemiluminescent signal produced by the GST-BAP protein. Active compounds (primary assay) that scored as negative in all three counterscreen assays were validated as RORα ligands.
Results
Screening Assay Validation
In an effort to identify natural RORα ligands, a cell-free high-throughput assay was developed, which was sufficiently robust and sensitive to detect RORα activity in partially purified fractions produced from raw plant extracts. Among the different assays tested, a pull-down assay that measures TIF2 protein recruitment on the immobilized RORα protein (
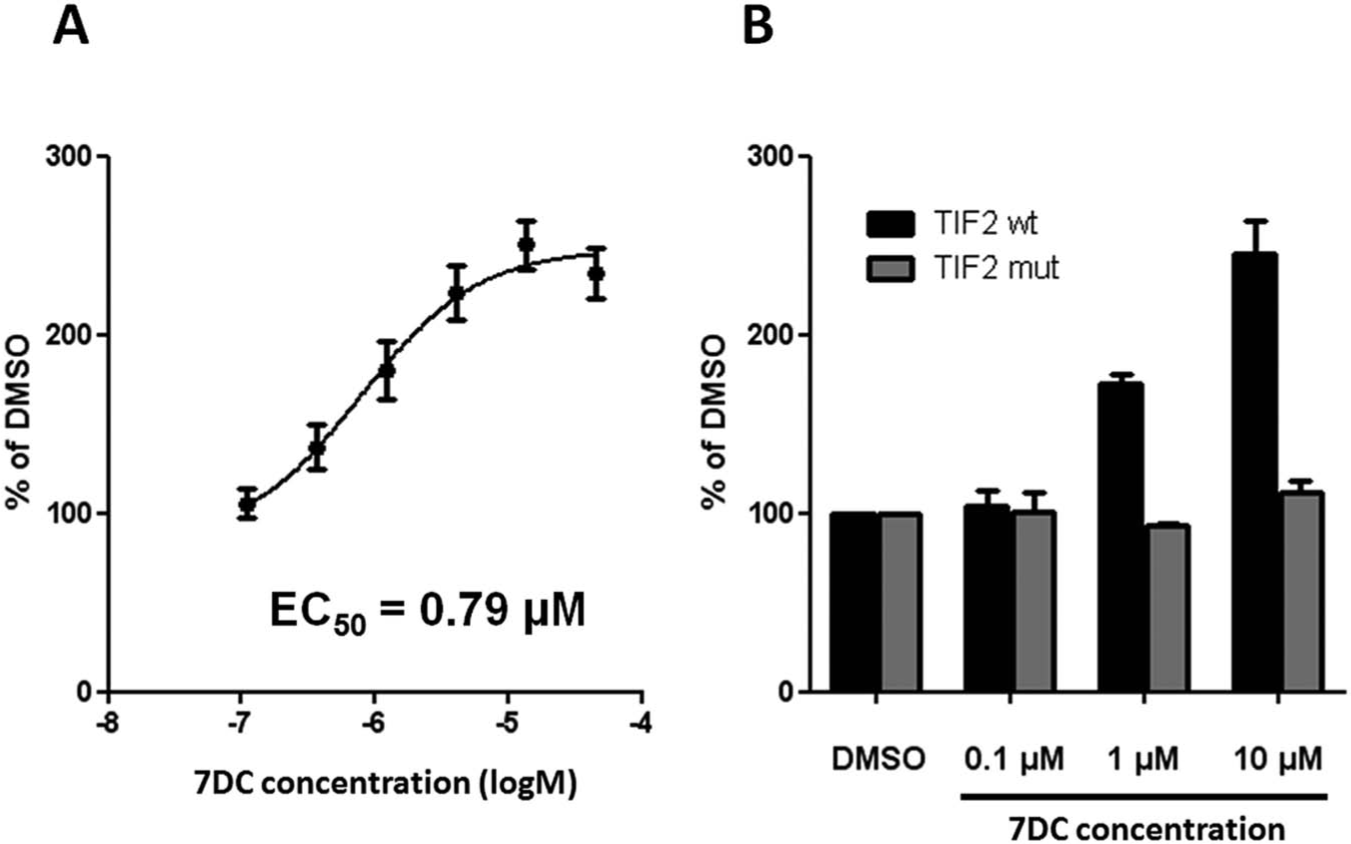
Development and validation of the primary screening assay. Purified GST-RORα protein was first immobilized on high-binding capacity streptavidin-coated plates. Upon several washes, a purified reporter protein (TIF2-BAP) and RORα reference agonist ligand, 7-dehydrocholesterol (7DC), were added. Binding reaction was performed overnight at 4 °C. A chemiluminescent substrate of the bacterial alkaline phosphatase (BAP) was then added upon extensive washes, and the signal was finally developed for 30 min at 37 °C in the dark. The reporter signal (relative light units [RLU]) was normalized to the basal signal, obtained for the solvent (DMSO). As shown in
Since the capacity for HTS was an objective, the pull-down assay was validated in the 384-well plate format, where induction with 7DC (10 µM) was on average 2.6-fold versus DMSO, and the Z′ factor values were between 0.5 and 0.7.
Plant Extract Library Screening Results
Once validated on steroid ligands, the pull-down assay was used for screening of the commercially available BILOBAC N library (Bicoll GmbH, München, Germany), which consists of 12,000 plant extract fractions (plant Profiles). These extracts were originally produced from 100 different and rare endemic Asian plant organs (leafs, stems, roots, etc.). For each plant, a set of 96 fractions originating from a nonpolar crude extract (obtained with dichloromethane [DCM] and ethyl acetate [EtOAc] extraction) and the corresponding set of 96 fractions of a more polar crude extract (obtained with EtOH extraction) were both submitted to screening. The complexity of the fractions prepared with this technology is considerably reduced as compared with the crude plant extract, and the fractionated material was easily amenable to high-throughput compatible procedures. Due to the fractionation procedure and the sequential screening of each fraction in the series, the signals measured in the neighboring wells form a continuous, bell-shaped distribution of the activity, resulting in active fraction clusters (
The initial screening was performed in simplicates at a concentration of 50 µg/mL. Fractions from three different plant fraction plates (96-well microplate format) were transferred onto one 384-well reaction plate, and the remaining empty wells were filled with either DMSO or the reference compound. The primary screen was performed as described in the Materials and Methods on 12,000 fractions, which correspond on average to between 36,000 and 120,000 different compounds.
20
To avoid a significant background activity, we have qualified only those clusters with an activity that was at least 2 × the standard deviation (SD) of the mean induction as calculated from all plates. Furthermore, only clusters with an activity of 40% or more, as compared with the induction obtained with the reference compound (7DC), were considered.
Primary screen data were analyzed manually to exclude all the active clusters that did not satisfy primary hit criteria. By applying this method, 18 potential agonist clusters were identified (
Fine Fractionation and Structure Elucidation
Fractions of the plant extracts (plant Profiles) represent a broad logP range. Empirical methods established that the logP-value extends with high reproducibility from 10 (A01 coordinate) to 1 (H12 coordinate) on each 96-well plate for different plant species (not shown). To ensure the structural diversity of compounds picked for further evaluation and to avoid any repeated selection of similar structures from different plants, active clusters from different logP regions were chosen for further study.
Typically, a deconvolution of an active cluster into a pure compound was achieved by preparative radial thin-layer chromatography (TLC) of the combined fractions on 1-mm silica gel plates. On average, one confirmed hit cluster led to a resubmission of between 5 and 20 partially purified compounds (purity from 70%–90%) for RORα activity evaluation. In the primary screen, the most active RORα agonist cluster, 8B6-MP-068E12-F7 (from the Agavaceae, Dracaena cambodiana), had a relative activity of approximately 60% of the reference compound. This activity was reconfirmed in a separate experiment and was specific for RORα, since no activity was observed in the assay that contained no RORα protein (
Fig. 2A
). Fine fractionation and isolation of active compound(s) of the cluster 8B6-MP-068E12-F7 were performed from the crude DCM and EtOAc extracts. The extract was obtained by refluxing dried plant material in the solvents, followed by filtration and evaporation. The crude extract was subjected to dry silica gel vacuum chromatography with a DCM-methanol gradient system (60:1 → 10:1). Compounds present in the active fractions were identified by silica gel TLC comparison with active plant Profiles (detection UV [254 nm], I2, Ce-Mo-phosphate). Compounds were enriched in the DCM-methanol = 25:1 fraction. Subsequent radial TLC on silica gel with the DCM-methanol gradient system (40:1 → 10:1) yielded 10 fine fractions for activity reconfirmation. The fine fraction 5 showed the highest activity, as determined by GST-RORα versus GST alone in the pull-down assay, with a relative activity of 310% as compared with DMSO (
Fig. 2B
). The major compound present in fraction 5 was chosen for structure elucidation studies. A final purification was conducted by a chromatography self-packed RP-18 column with acetonitrile/water, yielding 1.4 mg of pure desired compound with >90% purity (according to 1H NMR estimation). Structure elucidation was conducted as based on 1H NMR, 13C NMR, and 2D-COSY-NMR (coupled C, H-spectrum). The chemical shifts (1H NMR and 13C NMR) are consistent with those reported in the literature for (25S)-ruscogenin, which is a spirostan sapogenin compound (
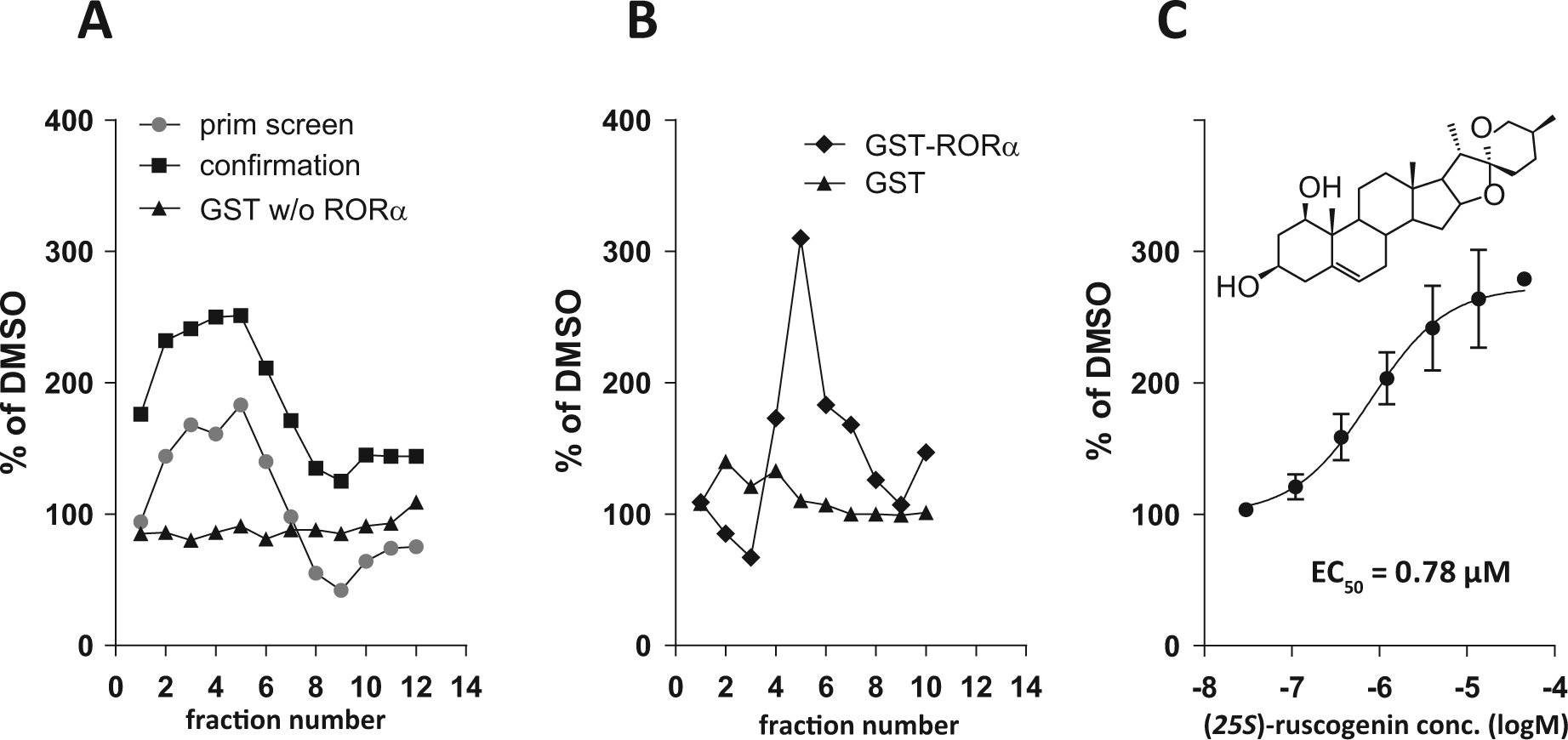
Identification and structural elucidation of a submicromolar RORα agonist ligand in the extract of the Agavaceae, Dracaena cambodiana. (
Identification of a Potent (25S)-Ruscogenin Analogue and Functional Properties In Vitro and In Vivo
Purification of milligram amounts of (25S)-ruscogenin suitable for extensive in vitro evaluation was very time and resource consuming. Therefore, some highly purified and commercially available (25S)-ruscogenin analogues were sourced for RORα agonist activity determination. Among the closest analogues, neoruscogenin (CAS #17676-33-4) appeared as the most potent RORα agonist compound (EC50 = 0.11 µM), as established by the pull-down assay ( Fig. 3A ). Furthermore, treatment with neoruscogenin, as described in detail in the supplementary materials and methods, significantly induced Gal4RE-luc reporter activity in COS7 cells that expressed Gal4(DBD)-RORα protein ( Fig. 3B ). In contrast, reporter activity was only minimally induced in COS7 cells that expressed the Gal4-DBD construct. The capacity of neoruscogenin to induce RORα target gene expression in the natural chromatin context was then demonstrated in HepG2 cells, where the expression of G6Pase and Bmal1, both well-established RORα target genes,14,18,22 was significantly induced upon neoruscogenin treatment ( Fig. 3C ). Detailed description of all experimental procedures is provided in the accompanying supplementary materials and methods.
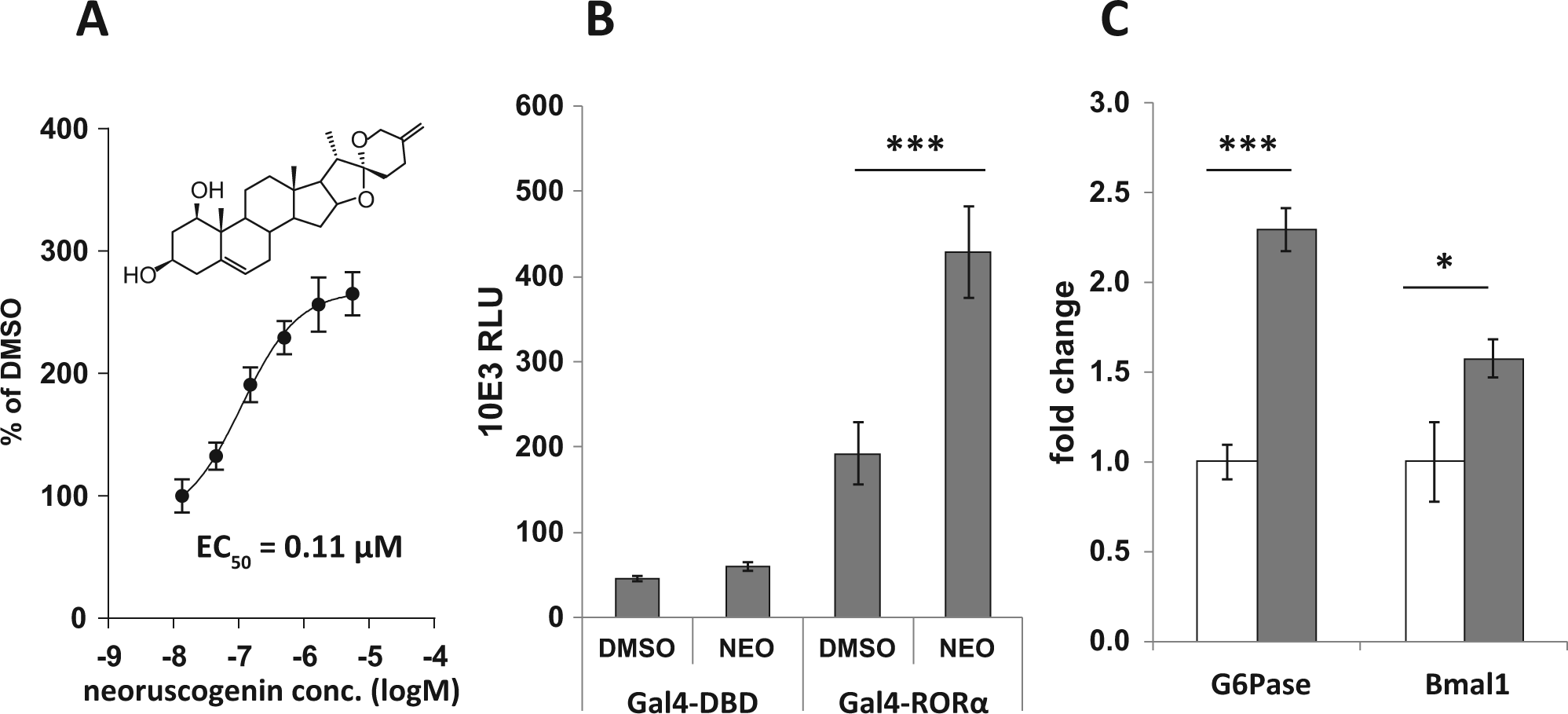
Structurally related neoruscogenin is a more potent RORα agonist as compared with (25S)-ruscogenin. (
Next, the selectivity, early ADME properties, and the pharmacokinetic (PK) profile of neoruscogenin were characterized before the compound was qualified for experiments in murine models. Neoruscogenin treatment did not significantly activate either steroid or xenobiotic nuclear receptors with the exception of a modest PXR activation (i.e., 1.8-fold reporter induction in cells that expressed GAL4(DBD)-PXR vs. 1.3-fold induction in cells that did not express the Gal4(DBD)-PXR) (
Neoruscogenin was metabolically stable, as 88% and 73% of the parent compound remained intact upon a 1-h incubation with mouse or human primary hepatic microsome preparations, respectively. Finally, classic PK studies were performed in mice (
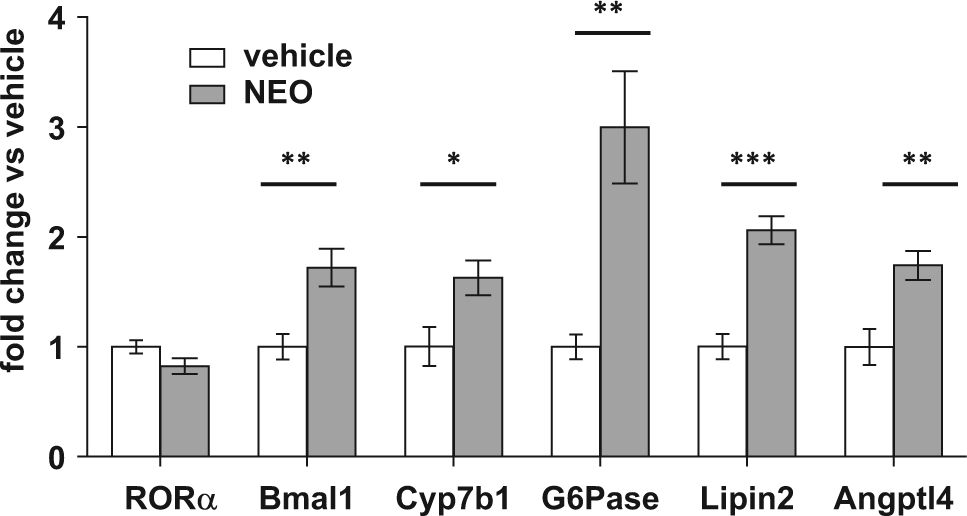
Neoruscogenin (NEO) activates hepatic RORα target gene expression. RNA was extracted and gene expression was measured by quantitative PCR in liver samples from mice that were treated with NEO (3 mg/kg/d, per os) for 7 days, as described in Materials and Methods. The expression of target genes was normalized to the expression of the 36B4 gene. Statistical evaluation: t test, *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
We describe in this article a straightforward and HTS-compatible procedure that enables the identification of naturally occurring ligands of orphan receptors in fractionated plant extracts. In principle, this approach is only limited by the technical difficulty to set up a robust cell-free assay, where the ligand binding domain of the target is exposed for interactions with preprocessed phytomaterial under nondenaturing conditions. Nuclear receptor RORα, a protein that has been targeted for more than a decade to identify potent and bioavailable agonists, was used here as an example of a challenging drug target.
In this work, roughly 12,000 readily available phytomaterial fractions (plant Profiles) were used, which should correspond to a library of up to 100,000 chemical entities. 20 In contrast, commercially available libraries of pure plant compounds contain only several thousand molecules. The technology used in the plant Profiles approach maximizes the chemical diversity present in the sample of 100 different Asian plant species used in this study. In contrast to crude extracts, which often interfere with biological assays to give false-positive readouts, our HTS procedure run on plant extract fractions gave reproducible and robust signals. Furthermore, positive fractions with RORα activity were further deconvoluted to obtain pure and active compounds within a period of 3 months. Classic problems associated with traditional extract testing, such as precipitation, interference with signal, and nonreproducible fractionation, are readily overcome by the process described in this article with no loss in product diversity. In conclusion, a preformatted (96-well), fractionated extract approach was chosen over a library of pure natural compounds to increase the structural diversity and avoid the burden of the purification of thousands of potentially inactive compounds.
The choice of a cell-free assay with a mechanism of action that relied on a naturally occurring protein-protein interaction (NR-LBD: NR-binding domain of a cofactor protein) was very important to obtain reproducible results and a significant ligand-induced response window (i.e., signal induction with the reference compound was ≥250% as compared with the basal signal). It was also taken into consideration during the assay development phase that there was a clear advantage of using a reporter assay that is based on chemiluminescence, to avoid interference by fluorescence that is naturally produced by plant metabolites.
The purification of (25S)-ruscogenin, which was the most active hit compound identified in this campaign, appeared difficult due to its low extract concentration and abundance of similar products. For practical reasons, the second part of this study concentrated on neoruscogenin (a close analogue of (25S)-ruscogenin), which is readily commercially available in gram quantities and more potent in terms of RORα activity. Indeed, the primary objective of this study was to rapidly obtain a pharmacological probe to be used for in vitro and in vivo validation studies.
Selectivity of compounds for a chosen target and the bioavailability in experimental rodent models are important selection parameters for high-quality pharmacological probes. Indeed, the selectivity of certain classes of natural compounds such as the flavonoids is quite low, and numerous mechanistically unrelated pharmacological activities have been described for such compounds. 27 On the contrary, we found that neoruscogenin did not show any significant effect on both RORβ and RORγ or on several other nuclear receptors that naturally interact with diverse steroid ligands.
The bioavailability and organ exposure to neoruscogenin in mouse were sufficient to obtain a clear effect on target-related hepatic gene expression. The effect on gene expression in the CNS was not examined, but judging from the PK data, brain tissue exposure (Cmax = 150-fold EC50) was sufficient to induce target gene regulation and confer pharmacologic action. In medicinal chemistry programs that start from small molecules derived from synthetic libraries, it can be challenging to obtain potent and bioavailable compounds within a couple of months. Therefore, we consider that the procedure described in this article is a real alternative solution when pharmacological in vivo proof of concept is needed to make educated decisions about an orphan target.
The results presented in this article validate two major strategic objectives. First, we have validated a robust procedure to identify potent and bioavailable pharmacological probes in complex plant extract fractions. Second, we demonstrated that carefully designed natural ligand identification procedures can rapidly deliver reliable chemical tools to characterize difficult orphan targets. Ruscogenins were identified among other biologically active compounds in extracts of Radix Ophiopogon japonicus and Ruscus aculeatus, plants that are known for their medicinal properties. Pure ruscogenin administration in a mouse model of pulmonary disease has favorable effects on hemodynamics, pulmonary vascular remodeling, and prevention of pulmonary hypertension development. 28 In line with these findings, ruscogenins showed anti-inflammatory,28,29 antithrombotic, 30 and antiapoptotic 28 activities in vitro. Future research will demonstrate which of these properties directly rely on RORα activation.
Footnotes
Acknowledgements
Bicoll members dedicate this article to Prof. Dr. Joachim Treusch, on the occasion of his 70th birthday.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by research grants from German Federal Ministry of Education and Research (BMBF, 0315012A and 0315944A) and by the EuroTransBio/OSEO grant No. PRESAGE 37149.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.