Glutathione transferases (GSTs) are cell detoxifiers involved in multiple drug resistance (MDR), hampering the effectiveness of certain anticancer drugs. To our knowledge, this is the first report on well-defined synthetic xanthones as GST inhibitors. Screening 18 xanthones revealed three derivatives bearing a bromomethyl and a methyl group (7) or two bromomethyl groups (8) or an aldehyde group (17), with high inhibition potency (>85%), manifested by low IC50 values (7: 1.59 ± 0.25 µM, 8: 5.30 ± 0.30 µM, and 17: 8.56 ± 0.14 µM) and a competitive modality of inhibition versus CDNB (Ki(7) = 0.76 ± 0.18 and Ki(17) = 1.69 ± 0.08 µM). Of them, derivative 17 readily inhibited hGSTA1-1 in colon cancer cell lysate (IC50 = 10.54 ± 2.41 µM). Furthermore, all three derivatives were cytotoxic to Caco-2 intact cells, with 17 being the least cytotoxic (LC50 = 151.3 ± 16.3 µM). The xanthone scaffold may be regarded as a pharmacophore for hGSTA1-1 and the three derivatives, especially 17, as potent precursors for the synthesis of new inhibitors and conjugate prodrugs for human GSTs.
Glutathione transferases (GSTs, EC 2.5.1.18) constitute an enzyme superfamily important in the medical and agricultural sciences because they are involved in cellular detoxification.1,2 GSTs confer protection to the cell against electrophilic metabolites, drugs, and toxic compounds. They do so by catalyzing the attack of the tripeptide glutathione (GSH) to the electrophilic center of such hydrophobic compounds, thus facilitating the degradation and transport of the respective conjugates. GST isoenzymes differ in their tissue specificity, expression, and distribution. Within mammals, GSTs are subdivided into classes on the basis of their amino acid sequence, for example, α, µ, π, σ, θ, ζ, κ, and ω.3,4 Human cytosolic GSTs are found as homodimers,3,4 with each monomer having an α/β domain that includes α1–α3 helices and a large α-helical domain composed of helices α4–α9. The former domain contains the GSH binding site (G-site) on top of the large α domain. A hydrophobic pocket (H-site) lies between the two domains in which the hydrophobic substrate binds and reacts with GSH.
Although GSTs’ detoxifying ability is a protective mechanism of the cell against deleterious compounds, it hampers, on the other hand, the effectiveness of certain chemotherapeutic drugs against cancer cells. This phenomenon leads to chemotherapeutic-resistant tumor cells that no longer respond appropriately to the applied therapeutic protocol. A possible origin for this problem appears to be an elevated expression of total GST activity in malignant tissue.5,6 For the GSTP1-1 isoenzyme, GST-dependent prevention of anticancer drug-induced apoptosis via direct interaction with signal transduction proteins has been suggested.7 Furthermore, homozygous hGSTA1*B breast cancer patients treated with cyclophosphamide and other chemotherapeutic drugs have a reduced death hazard during the first 5 years following diagnosis compared with homozygous hGSTA1*A individuals (hazard ratio, 0.3).7 This observation has been attributed to the detoxifying role of the isoenzyme hGSTA1 against therapeutic metabolites and/or the therapeutic drug.
Several principles have been proposed for overcoming multiple drug resistance (MDR) by GST overexpression, employing synthetic drugs and prodrugs as GST inhibitors,8,9 none of which has focused on synthetic xanthone analogues. Xanthones have several potential therapeutic applications such as antiviral, antimicrobial, anticancer, and antioxidant activity.10–12 The antibiotics bicaverin, cervinomycins A1 and A2, psorospermum, lysolypin, and citreamicins have a xanthone fragment in their architecture10,11 as well as naturally occurring bioactive compounds.13 Their planar structure confers DNA intercalation ability upon them, and hence expected cytotoxicity, which is considered a drawback in their use. Nonetheless, even with these limitations, their selective inhibitory power can still be optimized. An asset is that substituent effects provide important information on interaction energies during target binding, used in drug design and docking studies.
Many xanthone derivatives bearing methyl, hydroxy, methoxy, or phenyl groups or those possessing complex polycyclic frameworks have been isolated from families of higher plants, fungi, and lichens.10,11,14 Most reports are based on derivatives isolated from natural sources, thus hampering the availability of the material in sufficient quantity. Indeed, studies with xanthones as GST inhibitors have been performed only with compounds of natural origin.15–17 On the other hand, the synthesis of analogues from suitably substituted fragments may be and usually is a multistep process, detrimental to the overall yield of the required structure. Apparently, being able to introduce functional groups directly onto the xanthone pharmacophore scaffold (core structure) and perform transformations forthwith would be an approach of choice. This concept has been put to the test with a lithiation-electrophilic quench protocol, reported recently.18 The emerged reactivity profile has been used in the present investigation. In fact, to our knowledge, this is the first report on well-defined synthetic xanthones as potential inhibitors against the human isoenzyme GSTA1-1, known to be involved, together with hGSTP1-1, in MDR. Since any structure-activity relationship (SAR) study begins with the identification of a lead compound that shows promising inhibitory potency for the target enzyme, one may anticipate that xanthones identified in the present report as having high inhibitory potency could be exploited as lead structures.
Materials and Methods
The expression plasmid pOXO4-GSTA1 was a much appreciated gift from Prof. C. S. Morrow (Department of Biochemistry, Wake Forest University School of Medicine, Winston-Salem, NC). The human colon adenocarcinoma cell line Caco-2 was obtained from the American Type Culture Collection (Manassas, VA). Reagents were used as commercially purchased, whereas solvents were purified and dried according to standard procedures.
Synthesis of the Xanthone Derivatives
Details on the synthesis of the xanthone derivatives used in the present work were described in earlier work.18–20 The method followed is summarized in Supplemental Figure S1, whereas the derivatives tested and data on reaction conditions and yields are laid out in Supplemental Tables S1 and S2, respectively. A commentary on the synthesis of the xanthone derivatives is presented in the supplementary information.
Expression and Purification of Human Recombinant Isoenzyme Human GSTA1-1
Human GSTA1-1 (hGSTA1-1) expression and purification have been described by Koutsoumpli et al.,21 and a description is also presented in the supplementary information.
Enzyme Assays for Testing the Xanthone Derivatives as Inhibitors and Inactivators for hGSTA1-1
Enzyme Assay for Determining GSTA1-1 Activity
Determination of GST activity was performed by monitoring the formation of the conjugate formed between CDNB and the tripeptide GSH at 340 nm (ϵ = 9600 L mol−1 cm−1) at 25 °C. A total assay volume of 1 mL contained, typically, potassium phosphate buffer (100 mM, pH 6.5), 1 µmol CDNB (33 µL from a 30-mM solution in ethanol), and 2.5 µmol GSH (33 µL from a 75-mM aqueous solution). DMSO was also added (20 µL, in place of equal volume of buffer) only for control assays of inhibition experiments with the xanthone derivatives (see below). The mixture was incubated at 25 °C for 5 min, prior to adding the enzyme sample (20 µL purified enzyme or 5 µL Caco-2 cell lysate). Initial velocities were determined in triplicate and corrected for spontaneous reaction rates when necessary. One unit of enzyme activity is defined as the amount of enzyme that produces 1.0 µmol of product per minute at pH 6.5 at 25 °C.
Screening the Xanthone Derivatives as hGSTA1-1 Inhibitors
The GST assay described above was used for screening the xanthone derivatives (Suppl. Table S1) as possible hGSTA1-1 inhibitors. Specifically, a total assay volume of 1 mL contained potassium phosphate buffer (100 mM, pH 6.5), 1 µmol CDNB (prepared in ethanol), 2.5 µmol GSH (prepared in water), and 100 nmol xanthone derivative (20 µL from a 5-mM solution in DMSO). The mixture was incubated at 25 °C for 5 min prior to the addition of the enzyme (20 µL of purified enzyme, ~0.18 ∆A340/min). The observed rate was used to calculate the remaining activity (%), taking as 100% initial activity value the rate observed after replacing the xanthone derivative by an equal volume of DMSO.
Testing the Xanthone Derivatives as GSTA1-1 Inactivators
In a volume of 0.904 mL of a potassium phosphate buffer (100 mM, pH 6.5), a sample of pure enzyme (10 µL, ~0.09 ΔA340/min) and 2 nmol of the xanthone derivative (7, 8, 17; 20 µL from a 0.1-mM solution in DMSO) were added. The mixture was incubated at 25 °C for 30, 60, 120, and 240 s in separate experiments, prior to adding the substrates CDNB (33 µL, 1 µmol, prepared in ethanol) and GSH (33 µL, 2.5 µmol, prepared in water). The enzyme activity was monitored (340 nm) by the formation of the conjugate between CDNB and GSH.
Inhibition Studies with Purified hGSTA1-1 and Caco-2 Cell Lysate
Determination of IC50 Values for Inhibitors 7, 8, and 17
Initial velocities for the hGSTA1-1–catalyzed reaction with CDNB and GSH as substrates were measured at 25 °C in the presence of various concentrations of xanthone derivatives 7, 8, and 17. The reaction mixture contained, in a total assay volume of 1 mL, potassium phosphate buffer (100 M, pH 6.5), 1 µmol CDNB (prepared in ethanol), 2.5 µmol GSH (prepared in water), and up to 100 nmol xanthone derivative in DMSO (up to 2% in assay mixture). The mixture was incubated at 25 °C for 5 min prior to the addition of pure enzyme (20 µL) or cell lysate (5 µL). The observed rate was used to calculate the remaining activity (%), taking as 100% initial activity value the observed rate (~0.15 ∆A340/min and 0.08 ∆A340/min for the purified enzyme and the cell lysate, respectively) after replacing the xanthone derivative by an equal volume of DMSO. The IC50 values were determined from a graph depicting remaining GST activity (%) versus xanthone derivative concentration.
Kinetic Analysis of Inhibitors 7 and 17 and BSP Using CDNB as a Variable Substrate
Initial velocities for the hGSTA1-1–catalyzed reaction with CDNB as a variable substrate were determined in reaction mixtures of a total volume of 1 mL (25 °C) containing: potassium phosphate buffer (100 mM, pH 6.5), 2.5 µmol GSH, and different concentrations of CDNB (13.5–2100 µM) in the absence and presence of inhibitor 7 (1.0, 1.5, and 2.0 µM) or inhibitor 17 (0.5, 1.0, and 2.0 µM) or the control inhibitor BSP (5, 25, and 100 µM).
Kinetic Analysis of Inhibitors 7 and 17 Using GSH as a Variable Substrate
Initial velocities for the hGSTA1-1–catalyzed reaction with GSH as a variable substrate were determined in reaction mixtures of a total volume of 1 mL (25 °C): potassium phosphate buffer (100 mM, pH 6.5), 1 µmol CDNB, and different concentrations of GSH (40–2475 µM) in the absence and presence of inhibitor 7 (0.10, 0.25, and 0.50 µM) or inhibitor 17 (7, 10, and 13 µM).
The GraFit3 computer program (Erithacus Software Ltd, Staines, UK) was used throughout for producing kinetic graphs and determining apparent kinetic parameters/constants and IC50 values.
Caco-2 Cell Line Culture and Preparation of Cell Lysate
Cells were grown as monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM) from Biochrom (Cambridge, UK) supplemented with 10% v/v fetal bovine serum (FBS; GIBCO, Carlsbad, CA), 1% v/v penicillin-streptomycin solution (GIBCO), and 1% v/v L-glutamine (GIBCO). The cells were maintained at 37 °C in an incubator with 5% CO2. For the preparation of stock cell pellet and thereafter cell lysate, Caco-2 cells were cultured in 175-cm2 plastic flasks (Corning, Corning, NY) up to 60% confluence. To detach cells, the medium was removed from the flask, and the cells were washed with 1× phosphate-buffered saline (PBS) and trypsinized with a trypsin/EDTA solution (GIBCO) for 2 to 4 min at 37 °C. To inactivate trypsin, cells were washed with DMEM containing 10% FBS, and the cell clumps were broken through pipetting. Subsequently, the cells were washed 5 times with 1× PBS, divided in 3 equal portions, and stored as a cell pellet at −80 °C. In one of the tubes containing cell pellet, potassium phosphate buffer (100 mM, pH 6.5, 500 µL) was added and the tube shaken until a suspension formed. The tube was then placed in an ice bath and subjected to sonication (3 × 10 s with 30-s intervals, 20% amp.) for cell disintegration prior to being centrifuged (13,000 rpm, 5 min). The supernatant (cell lysate) was collected, tested for GSTA1-1 activity22 using the assay described earlier (“Enzyme Assay for Determining GSTA1-1 Activity”), and used for determination of IC50 values.
Cytotoxicity Experiments for Determining LC50 Values for Caco-2 Cells with Derivatives 7, 8, and 17 and Xanthone
Cytotoxicity was evaluated for Caco-2 cells using the MTT assay, which measures the ability of viable cells to reduce a soluble tetrazolium salt to an insoluble purple formazan precipitate.23 Caco-2 cells used for the MTT assay were seeded at a density 1.0 × 104 cells/well in 96-well plates and preincubated for 48 h in DMEM containing 10% FBS before the addition of xanthone derivatives. These were dissolved in 100% DMSO and then diluted with serum-free DMEM as culture medium to different concentrations and added to Caco-2 cells cultured in serum-free medium for an additional 24 h (the final percentage of DMSO in cultures was a limiting factor, especially for derivative 8, in which the maximum concentration tested had to be restricted to 20 µM with up to 2% DMSO in culture). After removal of the medium, each well was incubated with 0.5 mg/mL MTT (Sigma-Aldrich, St. Louis, MO) in DMEM serum-free medium at 37 °C for 3 h. At the end of the incubation period, the medium was removed and the intracellular formazan was solubilized with 200 µL DMSO and quantified by reading the absorbance at 550 nm on a microplate reader (Optimax; Molecular Devices, Sunnyvale, CA). Percentage of cell viability was calculated based on the absorbance measured relative to the absorbance of cells exposed to the negative control.
Modeling and Docking: In Silico Creation of the hGSTA1-1 Structure and Docking of the Xanthone Derivatives to the hGSTA1-1 Model
The structure of hGSTA1-1 in complex with ethacrynic acid and its glutathione conjugate (PDB code 1GSE) was processed using MGLTools 1.5.4 (Molecular Graphics Laboratory, La Jolla, CA).24 The tripeptide substrate glutathione was added to the protein PDBQT file. Docking of the xanthone derivatives to hGSTA1-1 was performed using AutoDock 4.0 (Molecular Graphics Laboratory).25 Forty different global molecular properties have been predicted for the tested xanthone derivatives using QikProp version 3.4 (Schrödinger, LLC, New York, NY).26,27 The AutoDock free-energy scoring function is based on a linear regression analysis, the AMBER force field,28 and a large set of diverse protein-ligand complexes with known binding constants. The Lamarckian genetic algorithm (LGA) search method with default parameters was used. Limits for 5,000,000 maximum energy evaluations and 50 docking runs were set. All ligands were treated as flexible. Docking results were both visually inspected and quantitatively evaluated based on the estimated free energy of binding (FEB) values and fitness parameters (LE, FQ) for docking to the hGSTA1-1 binding site (Suppl. Table S3). All figures depicting 3D models were created using PyMOL, version 1.4 (Schrödinger, LLC).
Results and Discussion
Chemistry of the Xanthone Derivatives
Looking at the structure of 1 (Suppl. Table S1), certain features are evident: (a) two benzene rings are fused to a γ-pyrone ring; (b) the benzene rings, being identical, impart symmetrical elements to the structure; (c) the tricyclic structure is planar; and (d) the carbonyl group and the ring oxygen atom are the major determinants of the reactivity profile of 1. The former, as its dimethyl ketal, facilitates attack at the peri-positions, through coordination. The latter, through its lone pair, activates C2 (or C7) as well as its peri-positions. During lithiation, there is a competition among the lithiated species, the base, and the attacking electrophile. It is not clear whether a prelithiation complex or a stabilizing lithium-substrate interaction at the transition state is involved.
Monomethylation occurs at C4, whereas dimethylation occupies C1/C5 or C4/C5 positions.18 Similarly, iodination (not chlorination or bromination) occurs at C1, C4, C1/C5, and C4/C5. An aldehyde group is attached solely at C4, but an amide group occupies C1, mainly and to a lesser extent C4 and both C1/C5 positions.
Radical bromination (NBS/AIBN [or Boc2O] in MeCN/CH2Cl2) on the C4 methyl group gives the corresponding mono-bromomethyl derivative 7 as the major product (in 42% yield), along with the dibromo-analogue 8 as the minor one (in 17% yield). Using the same conditions on the 4,5-dimethyl derivative 6, all three bromo-products are formed (their distribution in the reaction mixture was dependent on the substrate/NBS ratio, the optimum being 2:1, whereas the most suitable solvent was MeCN/CH2Cl2 1:2). Using the low solubility of the xanthone core structure, particularly in nonpolar solvents, to our advantage, the polarity of the reaction medium was reduced to favor the desired monobromo derivative.
Nuclear bromination of 1, on the other hand, at C2 (or C7) or dibromination at those positions can be effected through an electrophilic substitution reaction using a large excess of Br2 in refluxing acetic acid.22 The applied strenuous conditions are needed to overcome the deactivating effect of the carbonyl group. Monobromination to 10 is the major reaction (in ~74% yield), whereas ~20% of the dibromo derivative 13 is obtained as a minor product. On the other hand, applying Friedel-Crafts conditions (Br2/AlCl3), the two derivatives are obtained in a ~1:1 ratio. Furthermore, C-arylation (mono- and diarylation) is affected by Suzuki-Miyaura coupling at the brominated sites.19
The planarity and rigidity of 1 lend steady anchoring to its complex with hGSTA1-1, whereas its substituent arms, at positions C1, C2 (and/or C7), C4, and C5, in derivatives 4 to 20, steer them through at a preferable conformation and binding mode.
Interaction of hGSTA1-1 with the Xanthone Derivatives
Prior to proceeding with the inhibition studies, control experiments were implemented with our enzyme preparation using BSP as a known hGSTA1-1 inhibitor.29 In silico analysis predicted that BSP interacts at a noncatalytic binding site, allowing simultaneous binding of the substrate CDNB in the catalytic primary site (Suppl. Fig. S2a). In concert with earlier observations,29 this finding was confirmed by kinetic studies, using BSP as an inhibitor and CDNB as a variable substrate, demonstrating a noncompetitive modality of inhibition versus CDNB (Suppl. Fig. S2b). Under the same experimental conditions, benzylsulfonyl-GSH has shown a competitive modality of inhibition.30
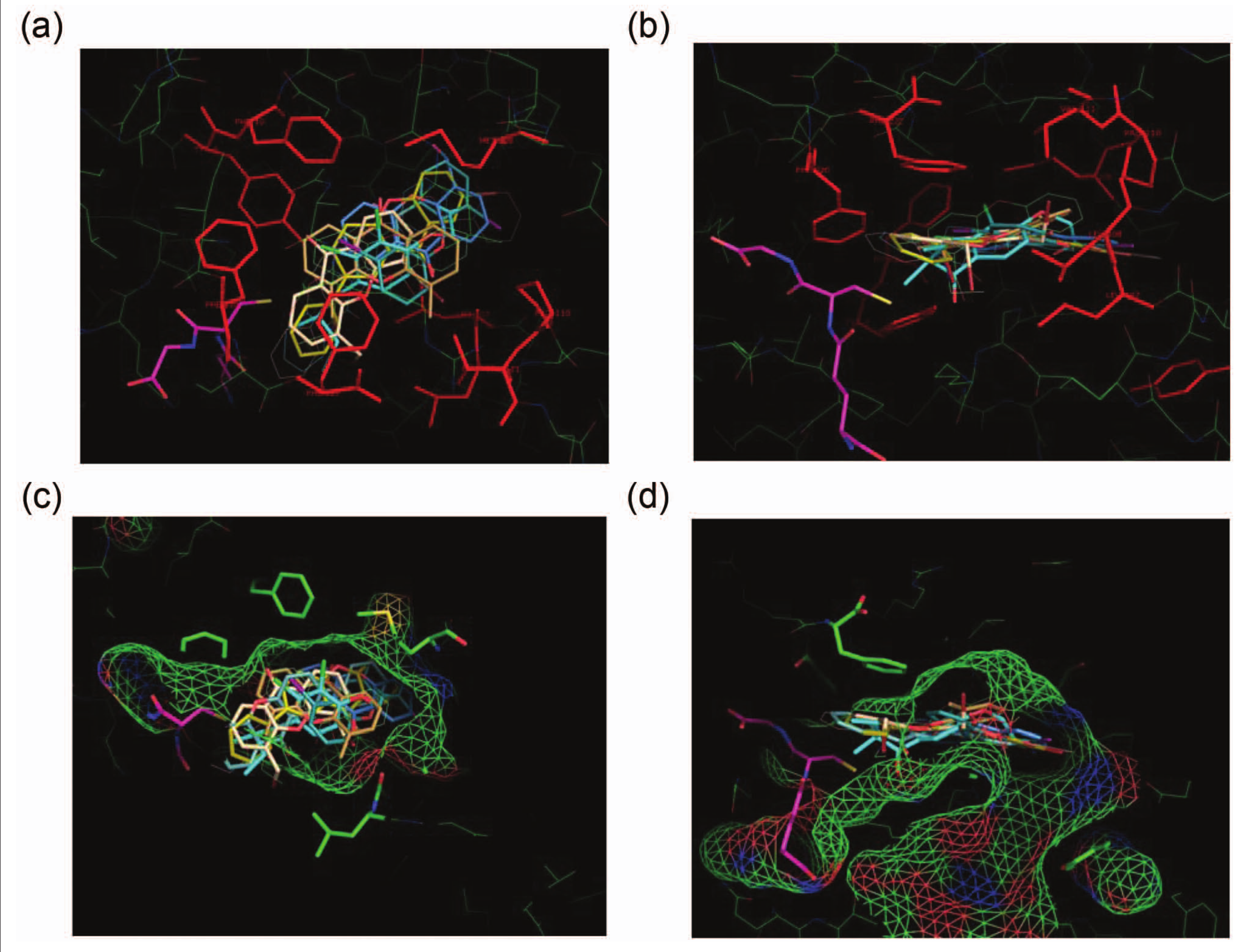
Looking at the inhibitory potency values (Suppl. Table S1 and Suppl. Fig. S3), certain variations are evident. Accordingly, at the lower range, methyl groups in 4, 5, and 6 appear to have a negligible effect, as does the amide in 19 and 20 or the monobromo derivative 10. Values for the monoiodo derivatives 14 and 15, on the other hand, are doubled, and a similar value is observed for the phenyl analogue 9 and a slightly lower value for the dibromo derivative 13. Notably higher values are those for 12 and 18. Analogues 11 and 16 show a further significant increase, whereas 7, 8, and 17 appear to top the list with an impressively high potency. It appears that these observations, marking not only actual changes but trends too, point to salient features of the substitution pattern and the nature of substituents in an aqueous environment, such as hydrophobic and H-bonding interactions.31 This is supported by the molecular modeling on the active site of hGSTA1-1 and the molecular mechanics–driven ligand docking. The low-energy positions for all derivatives indicate the optimum positioning of the ligands in a well-defined hydrophobic pocket surrounded by residues Tyr9, Phe10, Leu107, Leu108, Pro 110, Val111, Tyr166, Leu213, Phe220, and Phe222 (
Fig. 1a,b
), creating a hydrophobic cage (
Fig. 1c,d
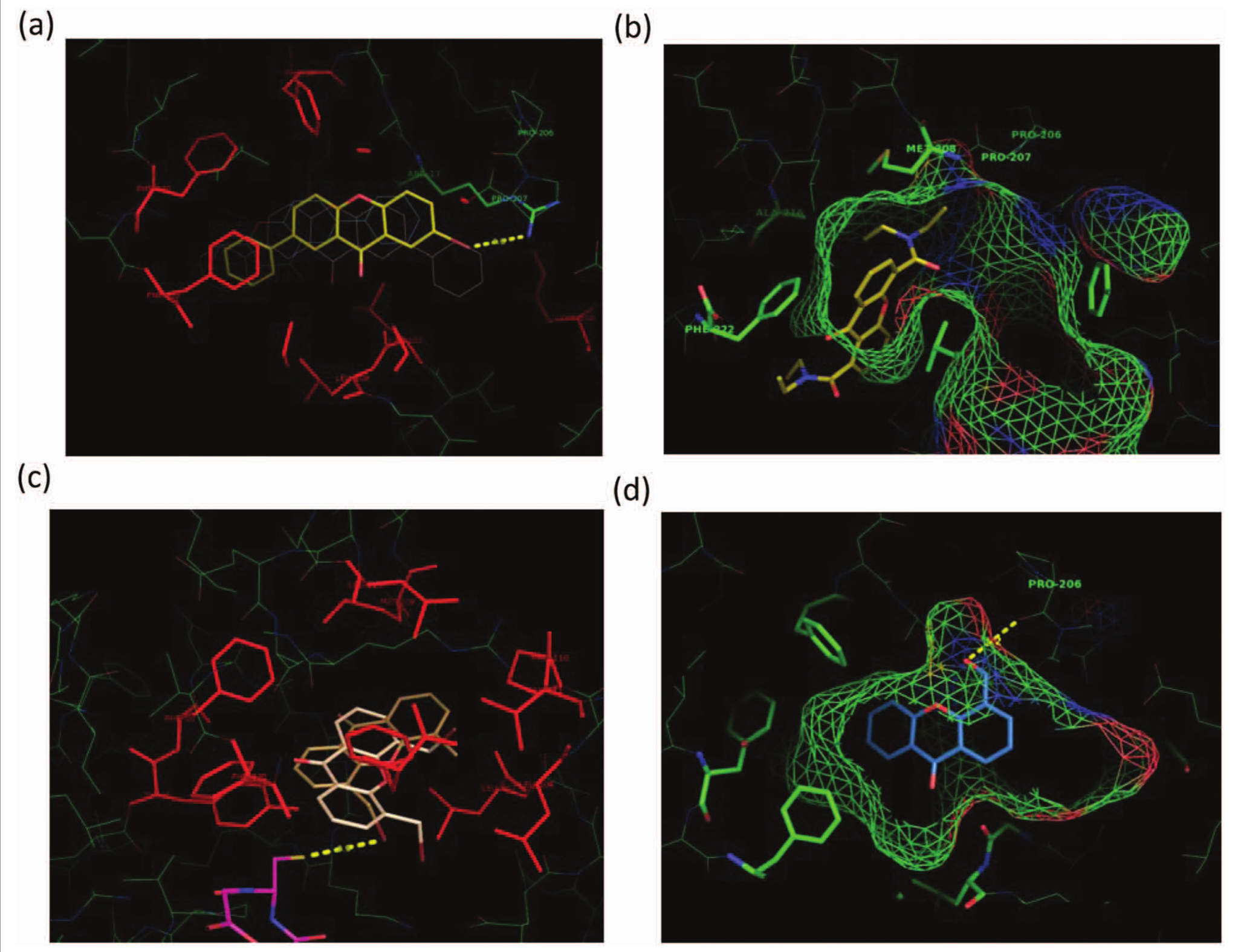
) that can readily accommodate the xanthone derivatives. In fact, 40 different global molecular properties have been predicted for the tested compounds using QikProp version 3.4.26,27 No strong correlation to potency has been identified so far, indicating that a more directional molecular description is required to link potency and structure. In addition to mono- and di-substituted compounds that have been synthesized and tested experimentally (Suppl. Table S1), 88 different structures have been generated and tested in silico with substitutions at C2 and C7 positions in addition to those at C4 and C5 by adding either or both polar/hydrophobic groups. Looking at the FEB values and fitness parameters (LE, FQ) for docking to the hGSTA1-1 binding site (Suppl. Table S3), it is apparent that all four substitution positions play an equally important role in the positioning of the three-ring structure (xanthone pharmacophore) to the binding pocket (Suppl. Fig. S4). Differences between di- and tetra-substituted compounds can be predicted as high as ~3.5 kcal/mol (Suppl. Table S3), indicating that tetra-substituted compounds can be good inhibitors as well. However, on the basis of the experimental findings, the most important positions appear to be C1, C4, and C5, peri- to the O-bearing pyran functionalities, at either mono- or di-substitution patterns. Furthermore, C1 is, in general, the position giving the highest values. Introduction of a second bromine (10 to 13) has a significant increase in inhibitory potency, whereas a similar addition of a phenyl group (9 to 12) has a weaker effect. In silico modeling has revealed that all phenyl substituted analogues at position C7 (9, 11, 12), when docked, form a π-π interaction with residue Phe222 (
Fig. 2a
). Worth noting, however, is the significant rise of 11 (42.4% inhibitory potency), indicating the dominance of substituent effects operating in concert, such as the hydrophobic character and planarity of the phenyl moiety and H-bonding with the halogen at a distance of 3.5 Å (11) from the guanidinium group of residue Arg13 (
Fig. 2a
). An earlier report32 endows the Arg residue with an important catalytic role. It appears that either hydrophobic or H-bonding interactions would lead to a tighter contact with the enzyme binding site, features that, when combined, would lead to a further substantial increase of the binding potency. Comparing 14 and 15 with 16 strongly suggests the prevalence of 1,5–di-substitution (16), at the peri-positions, over their mono-substituted variants (14, 15). This is supported by the clustering caused during docking where the iodine in 14 and 15 is placed in a very hydrophobic region, whereas in analogue 16, steric constraint forces iodine atoms into less hydrophobic positions. The notable increase observed in 18, as opposed to its congeners 19 and 20, is not clear. A lone pair–peri-repulsive interaction among the pyran carbonyl and amide substituent at C1 probably forces the latter to assume an out-of-plane orientation and become more available to intermolecular interactions with enzyme binding site. The peri-strain may also cause some ring σ-frame distortion. An intramolecular bidentate arrangement of the amide substituent at C4 or C5 with the pyran ring O lone pair toward a cooperative bonding (e.g., bifurcated H-bonding) with an enzyme amino acid residue creates a molecular volume not readily accommodated in the enzyme’s hydrophobic pocket (
Fig. 2b
, inhibitor 20), hampering alternative favorable intermolecular interactions. Accordingly, the inhibitory potency for 19 and 20, of similar magnitude, is low (6.7% and 7.1%, respectively; Suppl. Table S1). However, the amide group at C1 in 18, apparently through some peri–lone pair interaction with the carbonyl oxygen, may cause some ring distortion, facilitating the positioning of the structure in the enzymatic cavity. This may lend support to the measured significantly higher inhibitory potency (23.7%; Suppl. Table S1).
Low-energy conformations of the xanthone derivatives and glutathione (GSH) at the primary binding site of hGSTA1-1 as predicted by in silico molecular docking. All ligands are shown as stick renditions. GSH is depicted in magenta. S atoms are in yellow, N atoms in blue, and O atoms in red. Orthogonal views (a) and (b) showing enzyme amino acid residues (in red) forming a hydrophobic core around the binding site. Face (c) and side (d) views of the xanthone analogues in the enzyme binding site, which is depicted by a color chickenwire representation showing its volume, shape, and the polar characteristics. Green indicates hydrophobic regions, whereas red (–) and blue (+) indicate polar ones. The figure is created using the PyMOL program, version 1.5 (Schrödinger, LLC, New York, NY).
Low-energy conformations of four xanthone derivatives at the most probable binding position in the primary site of hGSTA1-1, as predicted by in silico molecular docking. All ligands are shown as stick renditions with S atoms in yellow, N atoms in blue, O atoms in red, and bromine atoms in brown. (a) Derivative 11 is shown in yellow, and the interaction of its bromine atom with the guanidiniun group of Arg13 (3.5 Å) is depicted with a yellow dotted line; the hydrophobic character and planarity of the phenyl moiety of 11 operate in concert with the halogen interaction, resulting in a tighter binding with the enzyme. (b) Derivative 20 occupies only part of the primary binding site of the enzyme depicted with chickenwire representation. Green indicates hydrophobic regions, whereas red (–) and blue (+) indicate polar ones. The created molecular volume of 20 is not readily accommodated in the enzyme’s hydrophobic pocket. (c) Derivatives 7 and 8 are shown in light brown and beige, respectively, whereas the cosubstrate glutathione (GSH) is depicted in magenta. The interaction of the bromine atom of derivative 7 with the sulfhydryl group of GSH (3.5 Å) is shown with a yellow dotted line. (d) Derivative 17 occupies part of the primary binding site of the enzyme depicted with a chickenwire representation. Green indicates hydrophobic regions, whereas red (–) and blue (+) indicate polar ones. The OH group (in red) of derivative 17 forms an H-bond interaction with the backbone carbonyl group of Pro206 (3.2 Å). The interaction is depicted by a dotted yellow line. All figures are created using the PyMOL program, version 1.5 (Schrödinger, LLC, New York, NY).
Identifying Xanthone “Lead Structures” for hGSTA1-1 Inhibitor Design
Derivatives 7, 8, and 17 are by far the most potent inhibitors for hGSTA1-1. Furthermore, they do not inactivate the enzyme after incubation at different times and in separate experiments (2 µM, pH 6.5, and 25 °C); reaction rates (ΔA340/min) obtained after incubating GST with compound 7 for 0, 30, 60, 120, and 240 s were 0.0152, 0.0150, 0.0150, 0.0153, and 0.0154, respectively.
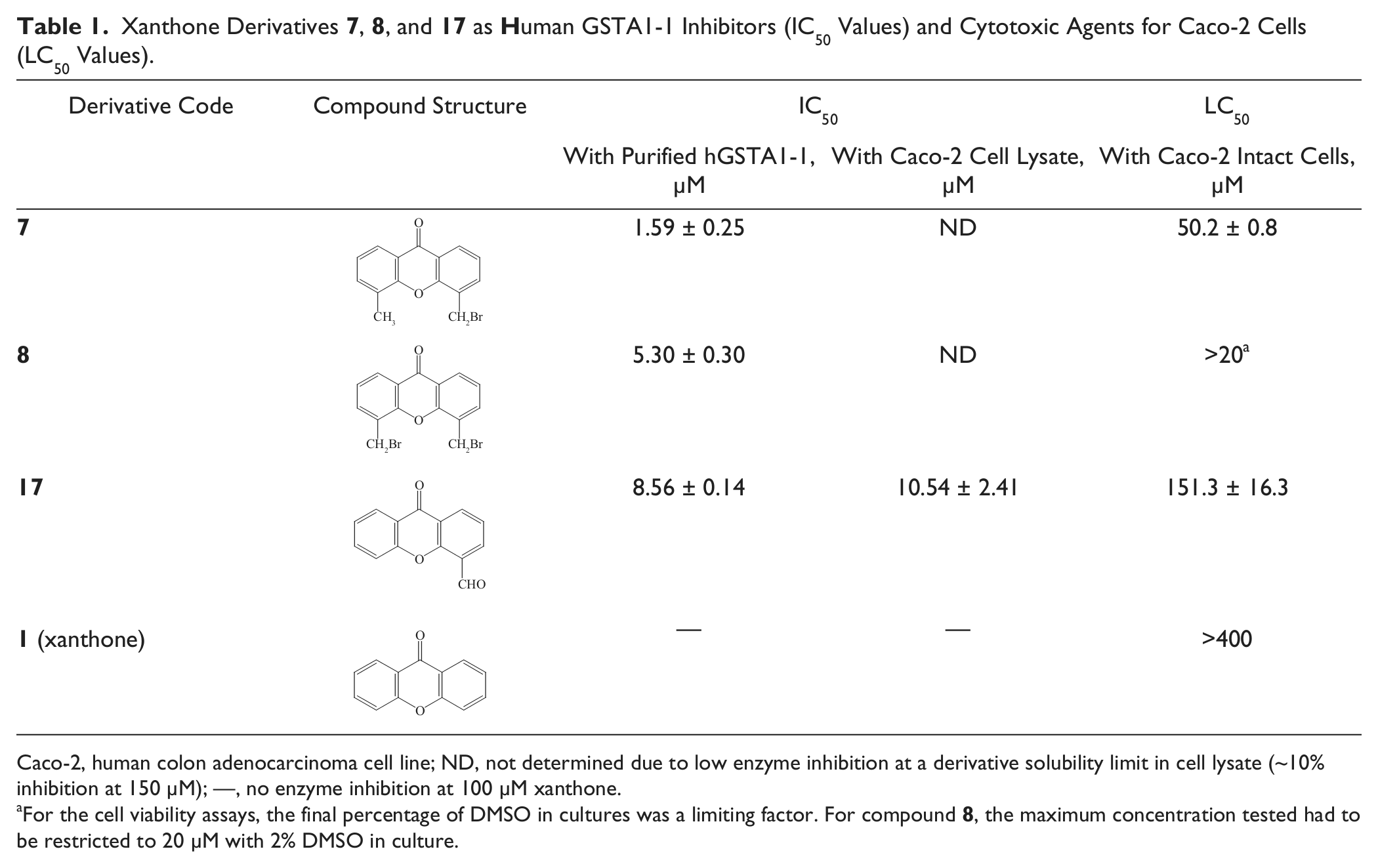
Derivatives 7 and 8 displayed high inhibitory potency (92.6% and 85.5%, respectively; Suppl. Table S1) and low IC50 values (1.59 ± 0.25 µM and 5.30 ± 0.30 µM, respectively;
Table 1
). In silico modeling analysis indicated that their low-energy docking positions place the xanthone moiety in the pocket in parallel with the benzene rings of Phe220, Phe222, and Tyr9 while the bromine is located 3.5 Å (7) (
Fig. 2c
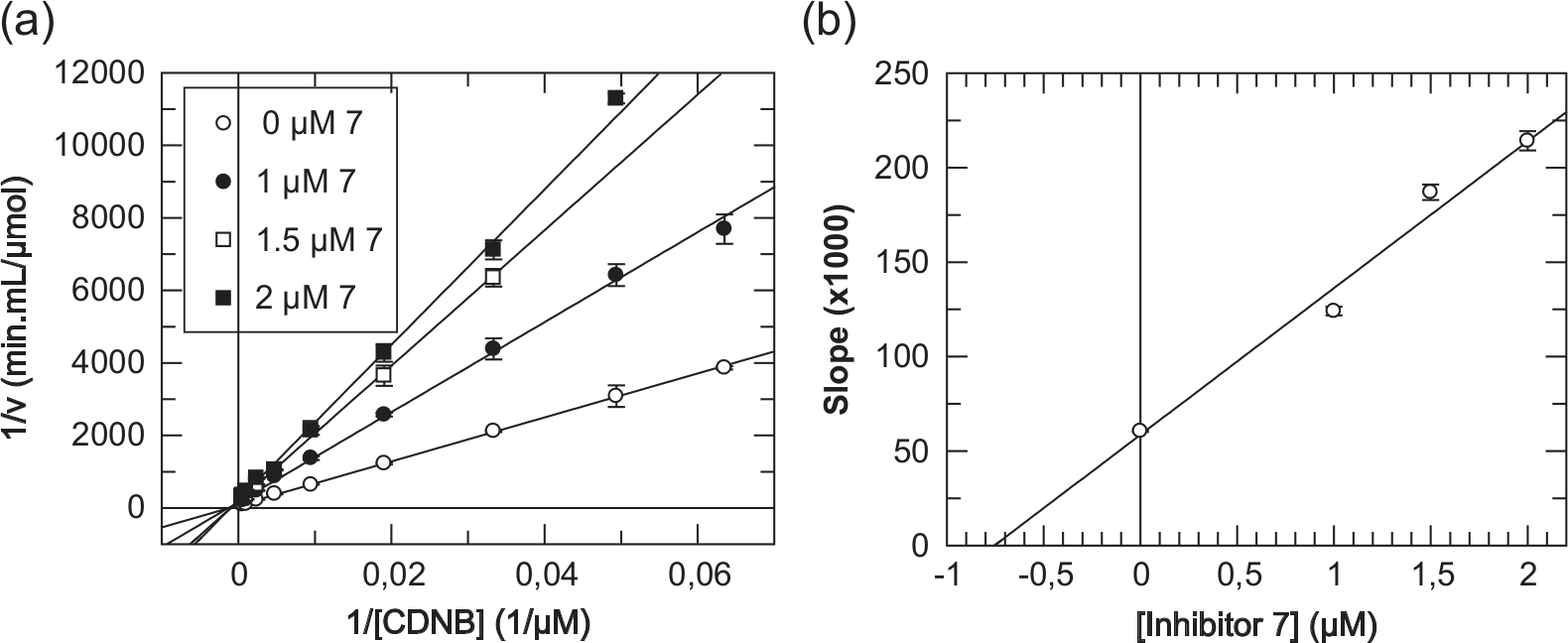
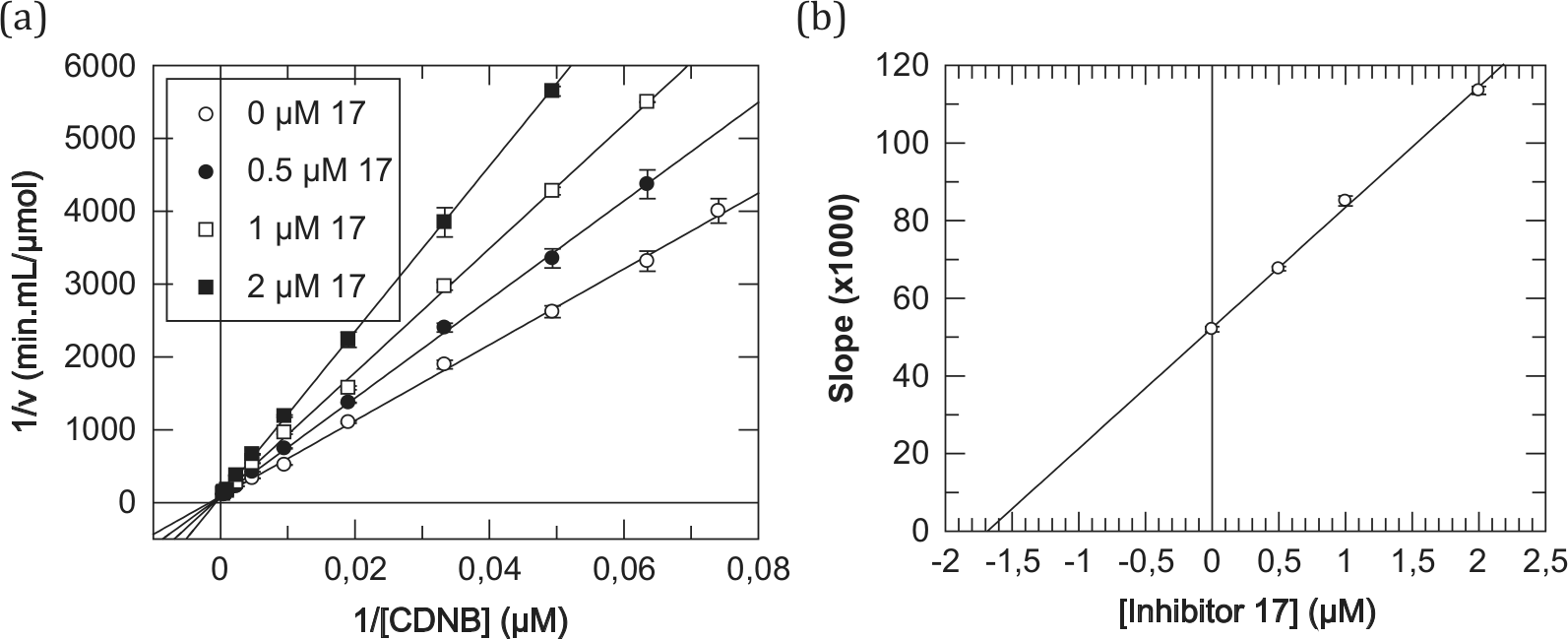
) and 5.1 Å (8) away from the sulfhydryl group of GSH and at an even longer distance (4.8 Å for 7 and 6.5 Å for 8) from the catalytically important Tyr9. This behavior is in coherence with the experimentally observed kinetic analysis. For example, derivative 7 displayed a purely competitive inhibition modality against CDNB as a variable substrate, on the basis of the linearity observed for both the double-reciprocal graph (
Fig. 3a
) and its secondary plot (
Fig. 3b
).33 With GSH, as a variable substrate, derivative 7 has shown a mixed (noncompetitive, α > 1) inhibition modality, on the basis of the lines of the double-reciprocal plot of initial velocities of hGSTA1-1 versus [GSH] intersecting to the left of the reciprocal velocity axis33 (Suppl. Fig. S5). This finding suggests binding of 7 at a site different from the GSH binding site, with that site being the catalytic CDNB binding site as shown earlier. In conclusion, derivative 7 competes with CDNB for the same binding site with a kinetically determined inhibition constant Ki(7) = 0.76 ± 0.18 µM. It is worth noting that although synthetic xanthone analogues have been reported in the past to affect antitumor activity,12,16,34,35 there has been no attempt to correlate experimentally such behavior with the inhibitory modality and strength of interaction versus human GST isoenzymes involved in MDR.
Xanthone Derivatives 7, 8, and 17 as Human GSTA1-1 Inhibitors (IC50 Values) and Cytotoxic Agents for Caco-2 Cells (LC50 Values).
Caco-2, human colon adenocarcinoma cell line; ND, not determined due to low enzyme inhibition at a derivative solubility limit in cell lysate (~10% inhibition at 150 µM); —, no enzyme inhibition at 100 µM xanthone.
For the cell viability assays, the final percentage of DMSO in cultures was a limiting factor. For compound 8, the maximum concentration tested had to be restricted to 20 µM with 2% DMSO in culture.
Purely competitive inhibition kinetics of hGSTA1-1 with derivative 7 using CDNB as a variable substrate. (a) Lineweaver-Burk (double-reciprocal) plot of initial velocities of hGSTA1-1 versus [CDNB] (13.5–2100 µM) at different concentrations of derivative 7 (0, 1.0, 1.5, and 2.0 µM). (b) Secondary plot derived from data of plot a. The inhibition constant Ki(7) for derivative 7 is the intercept on the inhibitor concentration axis. Points of enzyme velocity are the average of three enzyme assays. The plot is created using the GraFit3 program (Erithacus Software Ltd, Staines, UK).
We have implemented biological assays with the derivatives 7, 8, and 17 using cell lysate derived from human colon adenocarcinoma cells (Caco-2 cell line) and intact Caco-2 cells. With regard to derivatives 7 and 8, their enzyme inhibiting ability has been dramatically decreased when using cell lysate compared with purified hGSTA1-1 (
Table 1
). In fact, it has not been possible to calculate IC50 values for derivatives 7 and 8 since only ~10% enzyme inhibition was observed when approaching their solubility limit. This is probably due to interaction of the derivatives with various entities of the cell lysate, securing only a small free amount of the derivative for GST inhibition, indicative of low selectivity of 7 and 8 for GST. Furthermore, these derivatives functioned as cytotoxic agents with intact Caco-2 cells (Suppl. Fig. S6a,b). Especially derivative 7 showed a substantially lower LC50 value (50.2 ± 0.8 µM;
Table 1
) compared with plain xanthone (>400 µM; Suppl. Fig. S6d). Determination of an LC50 value was not possible for derivative 8 since the final percentage of DMSO in cultures was a limiting factor for the cell viability assays (Suppl. Fig. S6b). The derivatives tested were totally dissolved in 100% DMSO at different concentrations. Derivative 8 was the least soluble, showing complete solubility in DMSO only up to 1 mM. On the other hand, derivatives 7 and 17 and plain xanthone were completely dissolved in DMSO at 10, 25, and 50 mM, respectively. For derivative 8, the maximum concentration tested had to be restricted to 20 µM with 2% DMSO in culture.
Turning to derivative 17, its formyl group at C4 has no spatial restriction to any orientation of its carbonyl oxygen, being free to interact with the enzyme environment. The effective binding of derivative 17 to hGSTA1-1 is reflective of its high inhibitory potency (86.7%; Suppl. Table S1) and low IC50 value (8.56 ± 0.14 µM;
Table 1
). Furthermore, molecular modeling analysis indicated that 17 can be accommodated in the substrate site with the aldehyde group positioned in a polar extension pocket formed by the carbonyl oxygens of residues Pro206 and Pro207, so as to form an H-bond interaction (3.2 Å) with the backbone carbonyl group of Pro206 (
Fig. 2d
). This is in concert with kinetic analysis experiments, using CDNB as a variable substrate, according to which derivative 17 displayed a purely competitive inhibition modality on the basis of the linearity observed for both the double-reciprocal graph (
Fig. 4a
) and its secondary plot (
Fig. 4b
).33 With GSH as the variable substrate, a mixed (noncompetitive, α > 1) inhibition modality was observed with derivative 17 (Suppl. Fig. S7), as with 7 earlier. This behavior suggests that derivative 17 competes with CDNB for the same binding site, being a potent inhibitor for hGSTA1-1 with a kinetically determined inhibition constant Ki(17) = 1.69 ± 0.08 µM. The enzyme could not accept derivative 17 as a substrate since, in this case, the hemithioacetal formed incipiently as the cysteine S-conjugate of GSH is clearly transient,36 collapsing back to its individual components.
Purely competitive inhibition kinetics of hGSTA1-1 with derivative 17 using CDNB as a variable substrate. (a) Lineweaver-Burk (double-reciprocal) plot of initial velocities of hGSTA1-1 versus [CDNB] (13.5–2100 µM) at different concentrations of derivative 17 (0, 0.5, 1.0, and 2.0 µM). (b) Secondary plot derived from data of plot a. The inhibition constant Ki(17) for derivative 17 is the intercept on the inhibitor concentration axis. Points of enzyme velocity are the average of three enzyme assays. The plot is created using the GraFit3 program (Erithacus Software Ltd, Staines, UK).
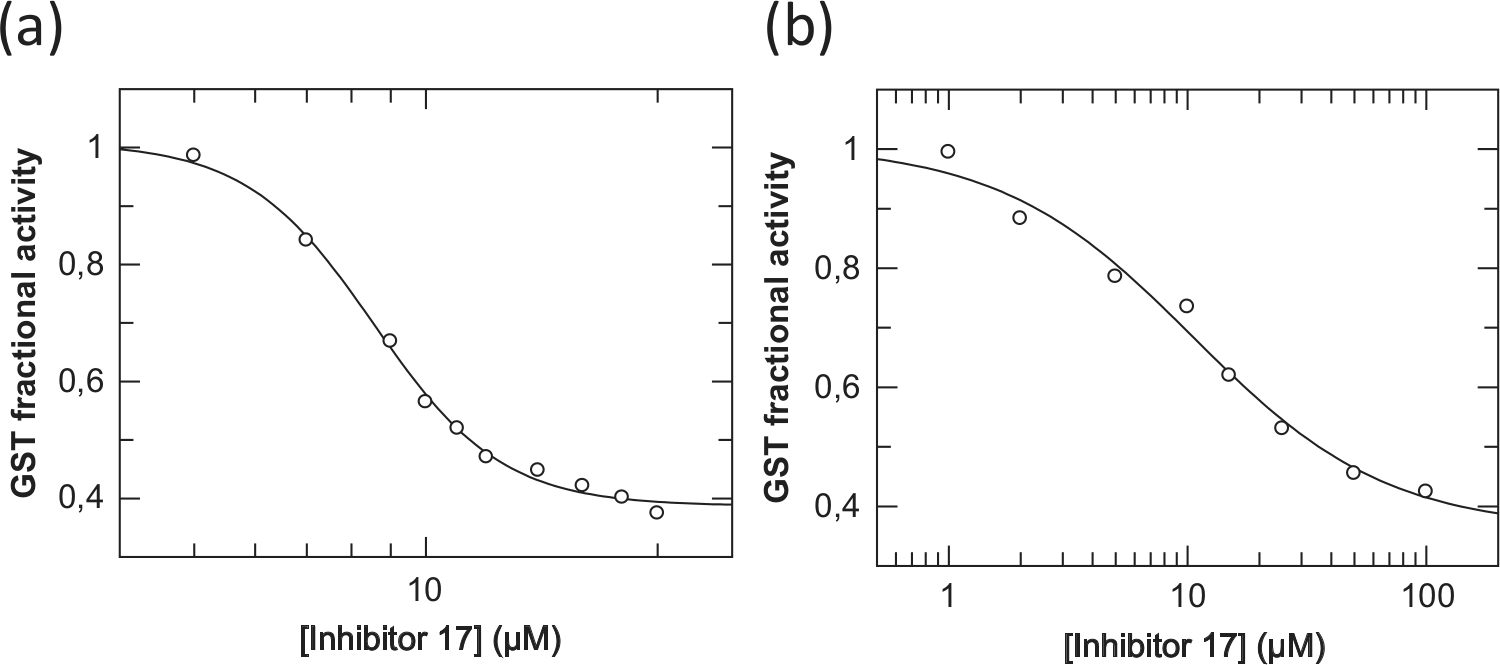
Testing derivative 17 with GST in the Caco-2 cell lysate, its inhibitory ability was similar to that with purified enzyme on the basis of the comparable IC50 values obtained: 8.56 ± 0.14 µM with purified hGSTA1-1 (
Fig. 5a
and
Table 1
) and 10.54 ± 2.41 µM with cell lysate (
Fig. 5b
and
Table 1
). The plateau observed at a high concentrations of derivative 17 may reflect phenomena of enzyme partial inhibition and/or solubility limitation of the inhibitor. It is reasonable to assume that the comparable IC50 values obtained for the purified and cell lysate enzyme suggest some degree of selectivity exhibited by 17 being not particularly prone to interaction with entities of the cell lysate, as did derivatives 7 and 8. Interestingly, derivative 17 has shown a substantially lower cytotoxic effect with intact Caco-2 cells (LC50(17) = 151.3 ± 16.3 µM; Suppl. Fig. S6c and
Table 1
) compared with derivative 7 (LC50(7) = 50.2 ± 0.8; Suppl. Fig. S6a and
Table 1
).
Concentration-response plots for determining IC50 values of the xanthone derivative 17 for hGSTA1-1 purified (a) and in Caco-2 cell lysate (b). The concentration values (µM) are presented on the logarithmic scale and the response values (as fractional activity of inhibited over uninhibited rates) on the glutathione transferase (GST) activity axis.
In conclusion, derivatives 7, 8, and 17 have shown a high inhibitory potency toward hGSTA1-1, of which derivative 17 was the only one to readily inhibit the enzyme in colon cancer cell lysate. Furthermore, all three derivatives were cytotoxic to Caco-2 cells, with 17 being the least cytotoxic. Thus, the xanthone scaffold may be regarded as a pharmacophore for hGSTA1-1 and the three derivatives, especially 17, as potent precursors for the synthesis of new inhibitors and conjugate prodrugs for human GSTs.30
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by the action THALES: “Glutathione transferases, multifunctional molecular tools in red and green biotechnology” falling under the Operational Programme “Education and Lifelong Learning” and is cofinanced by the European Social Fund and National Resources.
Supplementary material for this article is available on the Journal of Biomolecular Screening Web site at .
References
1.
WuB.DongD.Human Cytosolic Glutathione Transferases: Structure, Function, and Drug Discovery. Trends Pharma. Sci. 2012, 33, 656–668.
2.
ChronopoulouE.LabrouN.Glutathione Transferases: Emerging Multidisciplinary Tools in Red and Green Biotechnology. Rec. Pat. Biotech. 2009, 3, 211–223.
3.
FrovaC.Glutathione Transferases in the Genomics Era: New Insights and Perspectives. Biomol. Eng. 2006, 23, 149–169.
SargentJ. M.WilliamsonC.HallA. G.. Evidence for the Involvement of the Glutathione Pathway in Drug Resistance in AML. Exp. Med. Biol. 1999, 457, 205–209.
6.
KoderaY.IsobeK.YamauchiM.. Expression of Glutathione-S-Transferases alpha and pi in Gastric Cancer: A Correlation with Cisplatin Resistance. Cancer Chemother. Pharmacol. 1994, 34, 203–208.
7.
AdlerV.YinZ.FuchsZ. Y.. Regulation of JNK Signaling by GSTp. EMBO J. 1999, 18, 1321–1334.
8.
SauA.TrengoF. P.ValentinoF.. Glutathione Transferases and Development of New Principles to Overcome Drug Resistance. Arch. Biochem. Biophys. 2010, 500, 160–122.
9.
MahajanS.AtkinsW. M.The Chemistry and Biology of Inhibitors and Pro-drugs Targeted to Glutathione S-Transferases. Cell Mol. Life Scien. 2005, 62, 1221–1233.
10.
FotieJ.BohleD. S.Pharmacological and Biological Activities of Xanthones. Anti-Infective Agents Med. Chem. 2006, 5, 15–31.
11.
PintoM. M. M.SousaM. E.NascimentoM. S. J.Xanthone Derivatives: New Insights in Biological Activities. Curr. Med. Chem. 2005, 12, 2517–2538.
WangL.KuangL.PanX.. Isoalvaxanthone Inhibits Colon Cancer Cell Proliferation, Migration and Invasion through Inactivating Rac1 and AP-1. Int. J. Cancer2010, 127, 1220–1229.
17.
MukanganyamaS.BezabithM.RobertsM.. The Evaluation of Novel Natural Products as Inhibitors of Human Glutathione Transferase P1-1. J. Enz. Inhib. Med. Chem. 2011, 26, 460–467.
18.
Odrowaz-SypniewskiM. R.TsoungasP. G.VarvounisG.. Xanthone in Synthesis: A Reactivity Profile via Directed Lithiation of Its Dimethyl Ketal. Tetrahedron Lett. 2009, 50, 5981–5983.
19.
GardikisY.TsoungasP. G.PotamitisC.. Xanthones in Heterocyclic Synthesis: An Efficient Route for the Synthesis of C-3 O-hydroxyaryl Substituted 1,2-Benzisoxazoles and Their N-oxides, Potential Scaffolds for Angiotensin(II) Antagonist Hybrid Peptides. Heterocycles2011, 83, 1077–1091.
20.
GardikisY.TsoungasP. G.PotamitisC.. Xanthones in Heterocyclic Synthesis: An Efficient and General Route for the Synthesis of Regioselectively Substituted Phthalazines. Heterocycles2011, 83, 1291–1302.
21.
KoutsoumpliG. E.DimakiV. D.ThireouT. N.. Synthesis and Study of 2-(pyrrolesulfonylmethyl)-N-arylimines: A New Class of Inhibitors for Human Glutathione Transferase A1-1. J. Med. Chem. 2012, 55, 6802–6813.
22.
BeaumontP. O.MooreM. J.AhmadK.. Role of Glutathione S-Transferases in the Resistance of Human Colon Cancer Cell Lines to Doxorubicin1. Cancer Res. 1988, 58, 947–955.
23.
AlleyM. C.ScudieroD. A.MonksA.. Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1988, 48, 589–601.
24.
SannerM. F.A Programming Language for Software Integration and Development. J. Mol. Graphics Mod. 1999, 17, 57–61.
25.
MorrisG. M.GoodsellD. S.HallidayR. S.. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662.
26.
DuffyE. M.JorgensenW. L.Prediction of Properties from Simulations: Free Energies of Solvation in Hexadecane, Octanol, and Water. J. Am. Chem. Soc. 2000, 122, 2878–2888.
27.
JorgensenW. L.DuffyE. M.Prediction of Drug Stability from Monte Carlo Simulations. Bioorg. Med. Chem. Lett. 2000, 10, 1155–1158.
28.
CornellW. D.CieplakP.BailyC. I.. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197.
29.
KolobeD.SayedY.DirrH. W.Characterisation of Bromosulphophthalein Binding to Human Glutathione S-Transferase A1-1: Thermodynamics and Inhibition Kinetics. Biochem. J. 2004, 382, 703–709.
30.
AxarliI.LabrouN. E.PetrouC.. Sulphonamide-Based Bombesin Prodrug Analogues for Glutathione Transferase, Useful in Targeted Cancer Chemotherapy. Eur. J. Med. Chem. 2009, 44, 2009–2016.
31.
BissantzC.KuhnB.StahlM.A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084.
32.
DouradoD. F. A. R.FernandesP. A.MannervikB.. Glutathione Transferase A1-1: Catalytic Importance of Arginine 15. J. Phys. Chem. 2010, 114, 1690–1697.
33.
LeskovacV.Comprehensive Enzyme Kinetics; Kluwer Academic: New York, 2003, pp. 73–94.
34.
RewcastleG. W.AtwellG. J.BaguleyB. C.. Potential Antitumor Agents. 58. Synthesis and Structure-Activity Relationships of Substituted Xanthenone-4-Acetic Acids Active against the Colon 38 Tumor In Vivo. J. Med. Chem. 1989, 32, 793–799.
35.
RewcastleG. W.AtwellG. J.BaguleyB. C.. Potential Antitumor Agents: Structure-Activity Relationships for Side-Chain Analogues of the Colon 38 Active Agent 9-oxo-9H-xanthene-4-acetic Acid. J. Med. Chem. 1991, 34, 2864–2870.
36.
MiltonJ.BrandS.JonesM.F.. Enantioselective Enzymatic Synthesis of the Anti-viral Agent Lamivudine (3TC™). Tetrahedron Lett. 1995, 36, 6961–6964.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.