
Abstract
The TrkA-PathHunter cell-based assay was used in high-throughput screening (HTS) to identify compounds that inhibit nerve growth factor (NGF)/TrkA signaling. The assay was conducted in a 384-well format, and typical Z′ values during HTS ranged from 0.3 to 0.8. The reproducibility of IC50 values was good, and the use of cryopreserved cells was well tolerated, as judged by assay parameters such as Z′ and S/B and by comparison of IC50 values obtained with cells in culture. During hit deconvolution, TrkA-kinase inhibitors were identified with ATP-competitive as well as non–ATP-competitive mechanisms of action. Furthermore, other mechanisms of action such as NGF and TrkA antagonists were also identified. Because of the different molecular mechanisms identified, it is possible that subsequent optimization work to increase affinity and selectivity might lead to compounds that could have a better chance to evoke clinical efficacy without the adverse effects observed for nonselective TrkA inhibitors.
Introduction
Nerve growth factor (NGF) plays a key role in physiological as well as in pathophysiological processes in several human neuropathies, and it is one of several trophic factors that regulate the development and maintenance of the nervous system. The family of neurotrophins, of which NGF is one member, binds the tropomyosin-related kinase (Trk) family of receptor tyrosine kinases (TrkA, TrkB, and TrkC). 1 In addition, each neurotrophin also binds to the common p75 neurotrophin receptor (p75NTR), a member of the tumor necrosis factor receptor superfamily. 2 The functionality of neurotrophin receptors, Trks and p75NTR, is very complex. Apart from forming complexes, the receptors can also be transactivated in a neurotrophin-independent manner. 3 The complexity of neurotrophin receptor signaling is likely to account for the diverse neuronal and nonneuronal actions that neurotrophin exerts.
NGF has been demonstrated to be involved in several long-term trophic responses such as neuronal growth, survival, and differentiation, all classical responses to NGF. These effects of NGF have led to an interest to develop agonists of NGF/TrkA-signaling4–7 or of NGF itself as a therapeutic in neurodegenerative disorders such as Alzheimer disease.8–10 Apart from its long-acting neurotrophic effects, NGF also elicits rapid responses, including changes in neuronal excitability, synaptic transmission, membrane protein sorting, and neural plasticity.11–13 Many of these rapid actions are mediated through the effects of Trk receptors on ion channels, as exemplified by the effects on the sodium channel Nav1.7 (PN1).14,15
In addition to the original discovery of NGF as a trophic factor for neurons during development, it is now clear that NGF also plays an important role in acute and chronic pain conditions. 16 Thus, inhibitors of NGF or TrkA might provide new analgesic treatment. In line with this, several clinical studies with anti-NGF antibodies for the treatment of osteoarthritis have been performed. 17 Example of other diseases that could prove to be of interest for drugs that affect NGF signaling are chronic itch, 18 cancer, 19 and Alzheimer disease.8–10
There is a clear potential for small molecules that could intervene with NGF signaling without the drawbacks that are inherent for antibodies. In fact, there has been a long interest in developing inhibitors of TrkA, and several high-affinity inhibitors have been identified.19,20 Other approaches to interfere with NGF-TrkA signaling include small-molecule compounds such as ALE0540, 21 PD90780, 22 as well as cyclic peptide mimetics. 23 Several of the described TrkA inhibitors have been identified by the use of the intracellular kinase domain of TrkA in biochemical assays.20,24,25 Surprisingly, there are limited reports in which the use of a cell-based assay for high-throughput screening (HTS) purposes has been described.26,27 The use of cell-based assays provides a correct cellular environment, with its intrinsic high ATP concentration, which is a context well suited for identifying novel compounds affecting of NGF-TrkA signaling.
In this report, a pathway assay has been developed for HTS to identify compounds that affect NGF signaling. In addition, the development and use of assays necessary for deconvolution of the hits including a novel protein-protein interaction assay, surface plasmon resonance (SPR), nuclear magnetic resonance (NMR) spectroscopy, direct enzymatic assay, and NGF-induced neurite outgrowth are described.
Materials and Methods
Materials
U2OS-TrkA, U2OS-TrkB, and U2OS-Trk/p75 cells were from DiscoveRx (Birmingham, UK). Neuroscreen-1 cells (NS-1; #R04-0001-CI) were from Thermo Scientific Cellomics (UK). Eagle’s minimum essential medium (EMEM) ATCC #30-2003 were from LGC Standards AB (Borås, Sweden). Leibovitz s L15 medium, RPMI1640, fetal bovine serum (FBS), horse serum, penicillin/streptomycin, and Hoechst dye were purchased from Invitrogen (Carlsbad, CA). White poly-D-lysine plates (Biocoat #356661) and Imaging Collagen I–coated plates were from Becton-Dickinson (Franklin Lakes, NJ), and white assay 384-well plates were from Greiner (784201). Streptavidin labeled with europium-cryptate SA-K (610SAKLB) and polyclonal goat anti-human Fc labeled with XL665 (61HFCXLB) were from CisBio (Codolet, France). Recombinant human TrkA-Fc (175-TK-050) and anti-NGF antibody (MAB2561) were purchased from R&D systems. Potassium fluoride was from Scharlau (Barcelona, Spain); all other chemicals were from Sigma-Aldrich (St. Louis, MO).
TrkA PathHunter Cell-Based Assay
Cells were maintained subconfluent in EMEM supplemented with 10% FBS and penicillin/streptomycin. The final HTS protocol included detachment of the cells and a media change to EMEM containing 0.5% horse serum. Five thousand cells in suspension were thereafter seeded in poly-D-lysine–coated wells in a total volume of 20 µL by the use of a Multidrop Combi (ThermoLabsystem). Cell plates were thereafter left for 30 min and subsequently placed in cell incubators with a humidified atmosphere with 5% CO2 and incubated for approximately 18 h. On the following day, NGF was dispensed (14.5 µL of a 90 ng/mL solution) into wells of a low-volume 384-well plate (min-control wells received only buffer) by the use of a Multidrop. Cell plates were removed from the incubator and were, together with NGF and compound plates, placed in a BioCel1800 system. The experiment was initiated by the transfer of 0.5 µL of compound (in DMSO) from the compound plates to the NGF-containing wells. Max- and min-control wells received only DMSO. The plates were allowed to incubate for 20 min before 10 µL of NGF-compound mixture was aspirated from each well and dispensed into each well containing 20 µL of cells. This cell-NGF compound solution (final concentrations were 15 µM of compound and 30 ng/mL of NGF, respectively) was carefully mixed by repetitive gentle mixing steps. The plates were thereafter incubated for 3 h without sealing. After that, 15 µL of detection reagent was added to each well by the use of a Multidrop, and the plates were incubated for 60 to 90 min before luminescence was read.
In Vitro Biochemical TrkA Kinase Assay
The intracellular kinase domain of TrkA was supplied by Carna Biosciences (08-186). The ULight-TK peptide substrate and the LANCE Eu-W1024 anti-phosphotyrosine (PT66) antibody were provided by PerkinElmer (Waltham, MA). A total of 7.7 µL TrkA-solution (1 nM TrkA in 50 mM Tris-HCl, pH 7.5, 1 mM EGTA, 10 mM MgCl2, 0.01% Tween, 1 mM DTT, 50 nM ULight-TK) was mixed with 0.3 µL of compound in DMSO (final concentration, 15 µM). After a 15 min preincubation step, 2 µL of ATP was added to yield a final concentration of 100 µM. The reaction was run for 90 min at room temperature before it was stopped by addition of a 5 µL stop solution containing EDTA and detection antibody (final concentrations were 10 mM and 2 nM, respectively). Plates were thereafter incubated for 1 h at room temperature before read using an Envision reader (PerkinElmer) with an excitation wavelength of 337 nm and an emission wavelength of 665 nm.
Production of NGF Proteins
Recombinant protein production of NGF (Novoprotein, Shanghai, China) followed the approach as described in the literature. 28 In short, a synthetic gene, coding the human pro-domain (residues 19–121), followed by an engineered enterokinase cleavage site and the sequence of mature NGF, was subcloned into pET30a. Protein was expressed in Escherichia coli BL21(DE3) transformed with the resulting plasmid. Inclusion bodies were isolated, and pro-NGF was refolded in vitro under oxidizing conditions to allow for correct disulfide formation. Target protein was isolated from the refolding mixture using ion exchange chromatography, followed by enterokinase digestion to release mature NGF. The protein was subsequently purified on ion exchange chromatography with a final step on gel filtration chromatography (20 mM phosphate buffer pH 7.0 and 200 mM NaCl). NGF was eluted as a single symmetric peak at expected elution position for dimeric NGF. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis indicated that protein was more than 95% pure, and mass spectrometry (ms) confirmed correct mass of the 118 residue protein. The protein sequence of final product was SSSHPIFHRGEFSVCDSVSVWVGDKTTATDI KGKEVMVLGEVNINNSVFKQYFFETKCRDPNPVD SGCRGIDSKHWNSYCTTTHTFVKALTMDGKQAA WRFIRIDTACVCVLSRKAVR.
Protein at concentration >1 mg/mL was stored in gel filtration buffer in small aliquots at −80 °C. Several single-point mutants of human NGF and various orthologs of NGF were successfully produced in this manner. An avi-tagged form of human NGF was produced in which the final protein had an N-terminal extension MSGLNDIFEAQKIEWHE (referred to as avi-tagged NGF).
In Vitro Biotinylation of Avi-Tagged NGF
Purified avi-tagged NGF was biotinylated in vitro on the unique lysine in the N-terminal tag using a BIRA500 kit (www.avidity.com). In short, avi-tagged NGF (about 1 mg/mL) was buffer exchanged into 50 mM Tris pH 7.5 and 7.5 mM MgCl2 (one Roche complete inhibitor tablet EDTA-free per 50 mL) by gel filtration on a PD10 column. Biotin BirA ligase, biotin, and ATP were added (following kit instructions), and the reaction was incubated for 16 h at ambient temperature. The mixture was concentrated to >1 mg/mL followed by two subsequent gel filtration steps on NAP-5 columns in phosphate-buffered saline (PBS) to ensure complete removal of biotin in the final sample (referred to as Bio-NGF). Protein was more than 95% pure on SDS-PAGE, with >90% incorporation of a single biotin as confirmed by MS. Bio-NGF was stored in small aliquots at −80 °C before final use in assays.
In Vitro TrkA:NGF Protein-Protein Interaction Assay Using HTRF
TrkA-Fc (final concentration 0.84 µg/mL) and anti-Fc XL665-conjugated antibody (final concentration 3.8 µg/mL) were mixed in buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.05% bovine serum albumin [BSA], 0.8 M KF). A total of 10 µL/well of this mixture was added to each well in white 384-well assay plates using a Multidrop and incubated for 30 min. Bio-NGF (final concentration 0.065 µg/mL) was prepared in assay buffer with 0.1% BSA. Streptavidin 0.3 µg/mL labeled with Eu-cryptate (SA-K; final concentration 0.075 µg/mL) was prepared in assay buffer with 1.6 M KF. Added to all wells was 7.5 µL SA-K in low-volume Greiner plates using a Multidrop. Thereafter, test compound (0.3 µL) and 7.5 µL Bio-NGF was added to each well containing SA-K and incubated for 30 min. Ten microliters of the Bio-NGF:SA-K solution was thereafter transferred to the plate containing 10 µL/well of TrkA-Fc:anti-Fc antibody complexes to yield a final volume of 20 µL. After mixing, the plate was allowed to incubate for the indicated times. The fluorescence was subsequently analyzed using an EnVision reader at an excitation wavelength of 337 nm and an emission wavelength of 620 and 665 nm.
In Vitro TrkA:NGF Protein-Protein Interaction Assay Using SPR/BIACORE
The BIACORE assay for characterization of the TrkA:NGF interaction follows the protocol described by Abdiche et al. 29 The extracellular domain of TrkA (TrkA-Fc Chimera, Ala33-Glu407, R&D system, cat No. 175-TK) was coupled to a BIACORE CM5 chip using amine coupling chemistry. The interaction between NGF and TrkA was monitored as NGF was injected in the flow on to the immobilized TrkA. All experiments were performed in HBS-P buffer (GE Healthcare). The BIACORE CM5 surface was activated using a 100 µL injection of an EDC/NHS mix (GE Healthcare, amine coupling kit) at a flow rate of 5 µL/min. TrkA-Fc was diluted to 2 µg/mL in 10 mM acetate buffer pH 5.5 and immobilized until a level between 3000 and 8000 response units (RU). Finally, a pulse of 20 µL ethanolamine (GE Healthcare) was injected to deactivate the remaining reactive group on the sensor chip surface. Compounds were diluted in HBS-P buffer and preincubated with 70 nM NGF before injection onto the immobilized TrkA. Control samples with no added compound yielded the maximal binding response. Inhibition of the interaction was determined using the association slope at 10 s after injection start as binding response. The surface was regenerated with an injection of 5 µL 10 mM glycine pH 2.
Competition Binding Assay Using NMR
Purified 15N-labelled NGF was used to develop an NMR displacement assay using a reporter molecule. A sequential titration was conducted to determine the NGF affinity for HTS hits. First, an NMR spectrum is recorded at 100 µM reporter. NGF is then added to the sample to a final concentration of 0.5 µM, resulting in a signal reduction of approximately 50% for the reporter molecule. Subsequently, the compound of interest (10 mM in DMSO) is added to the sample in four steps, giving final concentrations of 2, 10, 50, and 250 µM, respectively. The affinity for the compound of interest is determined from a nonlinear least-squares fitting procedure of the six experimental data points (without and with protein and the four compounds of interest). The buffer was 50 mM d6-Tris pH 7.6 in D2O, and the experiments were run at 20 °C.
The data were recorded with 16 dummy transients and 64 acquired transients. A dual selective pulse (shape sinc1, 3 ms) was used in the excitation-sculpting scheme 30 to suppress both the residual water and DMSO signals simultaneously. A 200 ms CPMG relaxation filter was used with a 2 ms delay between the hard 180° pulses. The interscan delay was 3 s. The NMR experiments were conducted on a Bruker 600 MHz spectrometer equipped with a cryogenically cooled TXI-probe head. A SampleRail sample delivery system (Bruker BioSpin) connected the spectrometer with a Tecan sample preparation robot. 31 Each protein or ligand addition was done just prior to the NMR experiment. To avoid dilution with robot system liquid (water), an in-house constructed piston was used for mixing the sample in the NMR tube.
In Vitro Direct Binding to Biotinylated Avi-Tagged NGF Using SPR/BIACORE
A BIACORE Sensor Chip SA series-S (GE Healthcare) was prepared by immobilizing Bio-NGF using the biotin-streptavidin coupling procedure. Direct binding experiments were performed using the BIACORE 4000 instrument, in which compounds were injected in parallel and association and dissociation were monitored in real time. All experiments were performed in PBS buffer. In brief, Bio-NGF was diluted in PBS buffer to yield 4 µg/mL and injected at 10 µL/min for 5 min over the streptavidin surface on the Series S Sensor Chip SA until a response of 3000 RU was obtained. The capturing was followed by injection of 1 µM biotin (Sigma Aldrich) to block remaining empty binding sites on the streptavidin surface. Test compounds were injected in concentration series over the sensor chip surface with immobilized NGF for 1 min at a flow rate of 30 µL/min. The surface was regenerated with an injection of 5 µL 10 mM glycine pH 2.
Neurite Outgrowth Assay
Neuroscreen1-GFP (NS1-GFP) cells were cultured in RPMI1640 medium supplemented with 5% FBS and 10% horse serum containing 1 µg/mL puromycin in collagen I–coated flasks. Cells were seeded into 384-well BD-Imaging collagen I–coated plates in RPMI1640 medium containing 5% FBS and incubated with compounds in the presence of 6 ng/mL NGF for 3 d in humidified atmosphere with 5% CO2. Thereafter, the cell culture media were replaced by 20 µL of 4% paraformaldehyde containing Hoechst nuclear dye, and the cells were kept for 20 min at room temperature. After the simultaneous fixation and nuclear staining, plates were washed with PBS-Tween, and wells were then filled with PBS. The ImageXpressMicro high-content screening system (Molecular Devices, Sunnyvale, CA) was used for automated image acquisition, and image analysis was performed by MetaXpress software version 3.1 (Molecular Devices). The images were taken in the set of two wavelengths with 10× objective and four frames for each well and then analyzed with Neurite Outgrowth analysis module. Cells were identified based on Hoechst nuclear staining and neurites associated with cell bodies on GFP signal. Mean outgrowth per cell was used as a read-out, and, in addition, cell number was monitored to assess compound toxicity.
Results
Characterization of the TrkA PathHunter Cell-Based Assay
Briefly, the U2OS-cells overexpress two fusion proteins, that is, TrkA fused to a small peptide epitope (Prolink [PK]) of β-galactosidase (β-Gal) and SHC1 co-expressed with a larger sequence encoding a majority of β-Gal, termed enzyme acceptor. NGF binding to TrkA induces phosphorylation of the intracellular domain and hence recruitment of the SHC1 protein to the TrkA receptor via interaction with the phosphorylated amino acid residue Y490. The complementation between the small activating PK peptide on TrkA and the major part of β-Gal on SHC-1 leads to an active β-Gal enzyme. The activation of TrkA is quantified by measuring the amount of active β-Gal by its conversion of a nonluminescent substrate into a luminescent product (
To determine optimal assay conditions, the numbers of cells were titrated and the incubation time, temperature, and media were varied. The use of cryopreserved cells instead of cells in culture was also validated. The EC50 of NGF was 5.5 and 8.2 ng/mL for 2500 and 5000 cells, respectively (
Because NGF can bind to both TrkA and p75NTR (p75), the effect of co-expressing TrkA and p75 versus expression of TrkA alone was also studied. Parameters that were studied (pre- and post-HTS) included pharmacology with respect to ligand affinity and potency and efficacy of compounds. The finding by DiscoveRx that the correct pharmacology of neurotrophins was dependent on the presence of p75 was confirmed (data not shown). In contrast to the change in pharmacology of neurotrophins, the results in Figure 1 demonstrate that the affinity of inhibitors was not affected by the presence of p75, thus suggesting that these compounds did not bind p75. One interesting feature was that the maximal inhibition did not reach more than 100% in TrkA+p75 cells, whereas in cells overexpressing only TrkA, the inhibition could reach >100% ( Fig. 1b ). This is probably due to a different basal activity of the TrkA receptor in these two cell lines, where the TrkA cells have a higher basal activity than the TrkA+p75 cells. Cells expressing only TrkA were chosen for all further studies. The compounds used for this experiment were chosen based on known pharmacology (i.e., kinase inhibitors, compounds described in the literature to be known to interfere with the NGF-TrkA pathway or compounds identified during the pre-HTS). However, compounds acting through other mechanism could still be dependent on p75.
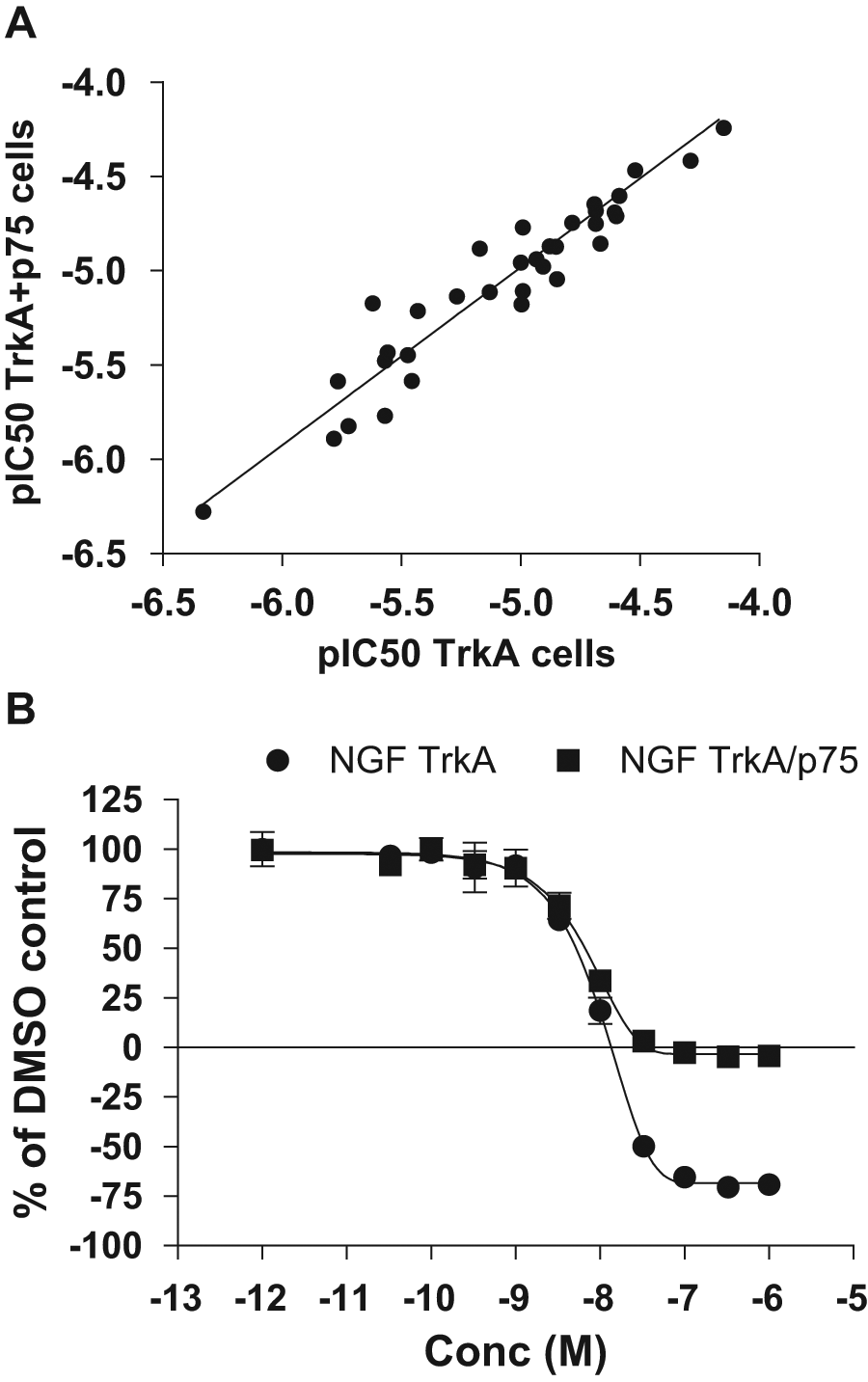
Comparison of TrkA and TrkA+p75 cells with respect to inhibition. Effects of inhibitors on U2OS-TrkA or U2OS-TrkA/p75 were examined by dose-response curves of compounds using 5000 cells and an incubation time of 3 h. (
Validation of the TrkA PathHunter Cell-Based Assay and HTS
The final assay conditions that were chosen for HTS included 5000 cells per well grown in 384-well poly-D-lysine–coated white plates, 30 ng/mL NGF (approximately EC90), and a final incubation time of 3 h at ambient temperature in the presence of 0.5% DMSO. The validated and optimized assay was fully automated and calibrated on a BioCel1800 system (data not shown). As shown in Figure 2a , the reproducibility of the automated assay with respect to IC50 determinations was good with an r2 = 0.82. In fact, when using an internally developed statistical tool to address assay variability and when analyzing a large number of compounds analyzed in at least two different experiments, we conclude that a 2.4-fold difference in IC50 value could be considered as statistically different (data not shown). The performance of a previously identified AstraZeneca TrkA kinase inhibitor (data not shown) was also monitored on each plate, and the descriptive histogram is illustrated in Figure 2b . The pIC50 was −8.22 ± 0.21 (mean ± SD, n = 117). The Z′ value for each plate was calculated according to Zhang et al., 32 and the mean value was 0.49 ± 0.09 (mean ± SD, n = 117). Furthermore, the use of cryopreserved cells in the assay was very well tolerated, as demonstrated by Z′ values >0.4 and a very good correlation of IC50 values between cells in culture and cryopreserved cells (data not shown).
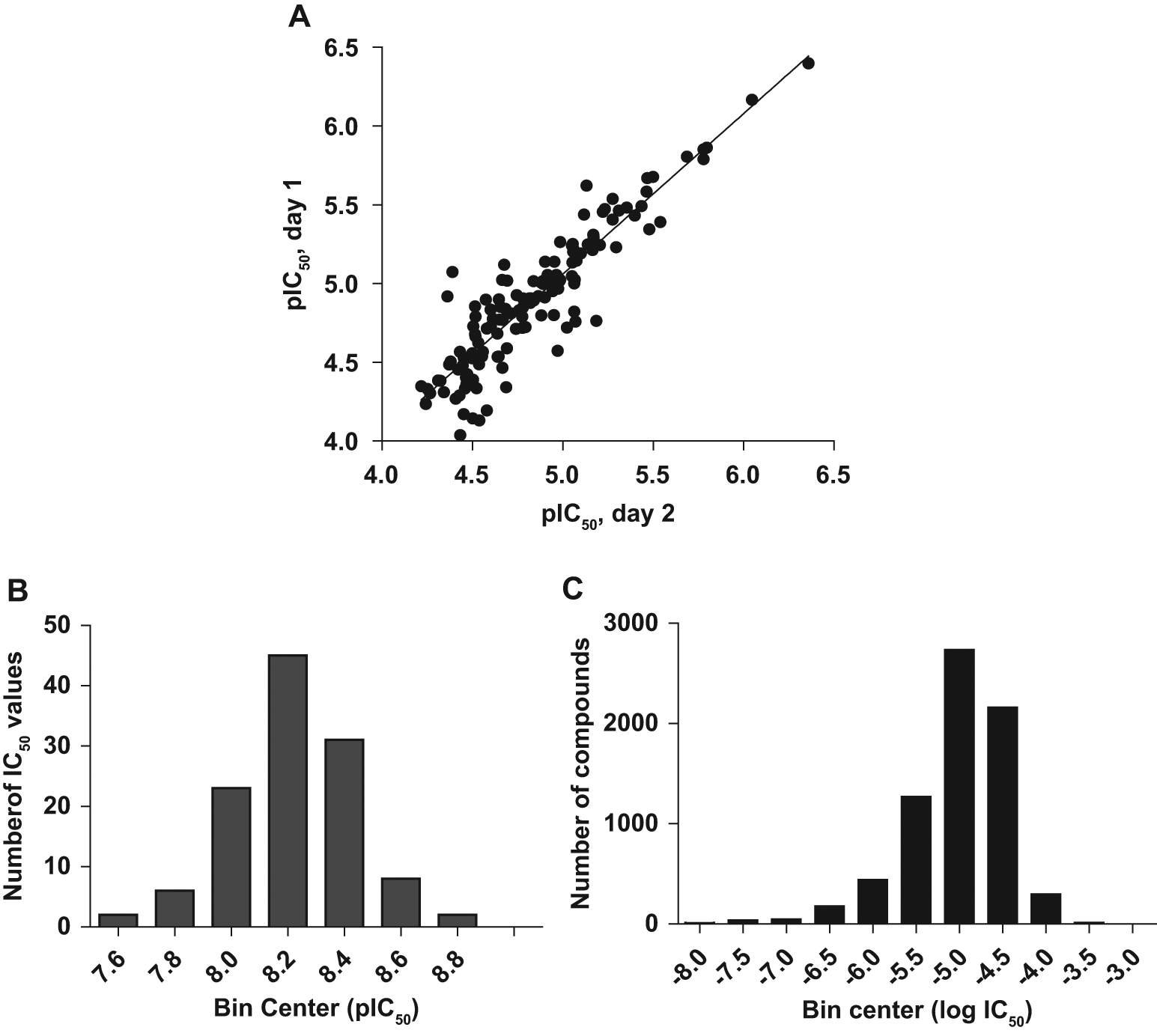
Comparison pIC50 variability and performance of a reference compound. Dose-response curves were generated using 5000 cells and an incubation time of 3 h. The mean values (± range; n = 2) were used in the nonlinear regression curve fit to generate IC50 values.
After an initial pre-HTS using 3765 inhibitor-like compounds and 26 14;058 diversity-representative compounds selected from the compound library, the optimized assay with cryopreserved cells was used to screen a part of AstraZeneca’s corporate compound collection at a concentration of 15 µM, and more than 677 14;000 compounds were screened. The Z′ factors of daily control plates during HTS were in the 0.3 to 0.8 range. A hit was defined as a compound with more than 40% inhibition, and the hit rate in the primary screen was 1.9%. The 13 14;436 active compounds were retested in triplicate, and the number of false-positives was only 2.5%, thus verifying the good reproducibility of the assay in HTS mode (data not shown). To determine potency, the IC50 values were determined for a total of 7172 compounds, and the frequency distribution is demonstrated as a histogram in Figure 2C .
Characterization of Hits Acting as TrkA Kinase Inhibitors
To identify the mechanism of action for the hits, we characterized 10 14;897 compounds that were available as stock solutions in a biochemical TrkA kinase assay. A limited assay development was conducted to establish basic assay conditions including concentrations of enzyme, peptide substrate, and ATP (data not shown). The final assay conditions included 2 nM of enzyme, 50 nM of peptide substrate, and 100 µM of ATP. Using these conditions, we demonstrated that the reaction was linear up to at least 2 h. The apparent Km (Km app) for ATP was 100 µM (data not shown). The Z′ of the final assay in automated mode was 0.78 ± 0.03 (mean ± SD, n = 6). We used the TrkA kinase assay to counterscreen compounds that demonstrated >40% inhibition in the TrkA PathHunter cell-based assay. Figure 3 demonstrates that a total of 6974 compounds were active in the TrkA kinase assay. After applying a soft filter for frequent hitters, 6230 of the compounds showed >50% inhibition in both the PathHunter cell-based assay and the TrkA kinase counterscreen.
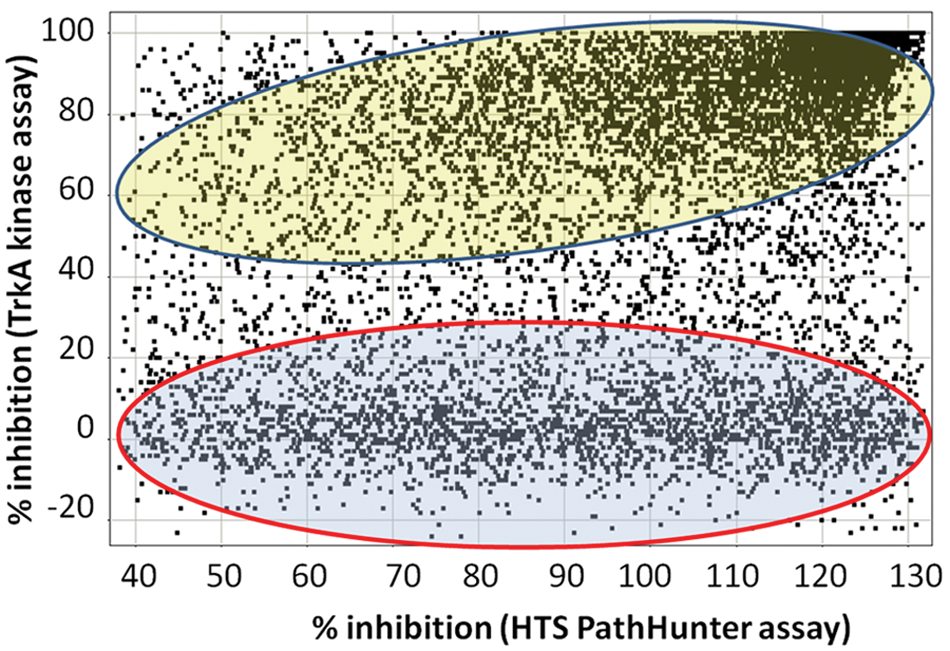
Comparison between TrkA PathHunter cell-based assay and TrkA kinase assays. Compounds were screened at 15 µM in both the TrkA PathHunter cell-based assay and in the biochemical TrkA kinase assay. The results from the PathHunter assay were compared against results from the TrkA kinase assay. The compounds in the blue area encircled by red have low activity in the biochemical TrkA kinase assay but high activity in the cell-based assay. In contrast, the compounds in the yellow area have high activity in the biochemical assay but low activity in the cell-based assay. The results are represented as percentage inhibition at 15 µM from single-point screens.
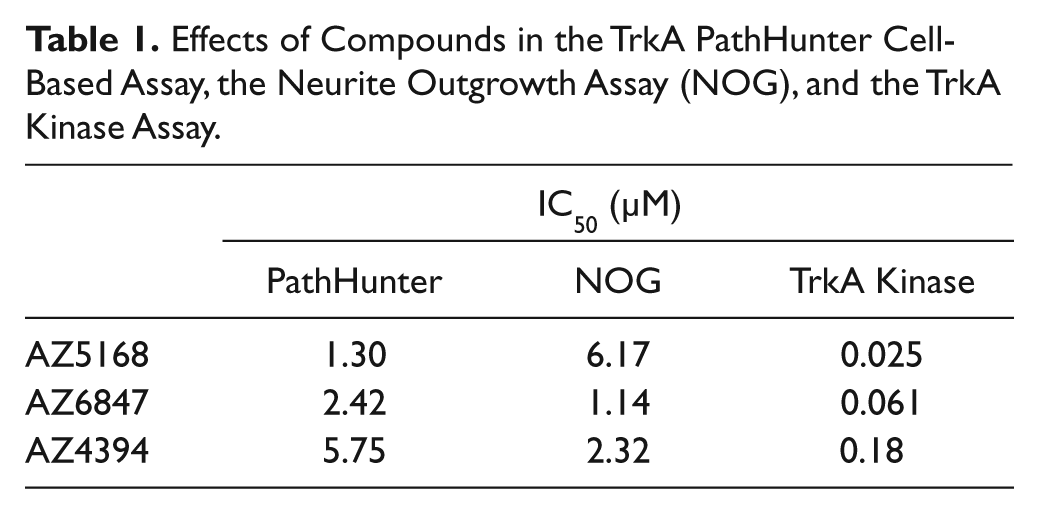
To identify TrkA kinase inhibitors that potentially could act in a non-ATP–competitive manner, 6411 compounds were rescreened that demonstrated >50% inhibition in the first TrkA kinase assay that was performed at low ATP concentration (100 µM). The second screen was performed at high ATP concentration (2.5 mM). The results from this second screen yielded >1900 actives. After applying a filter for frequent kinase hitters, the numbers of actives were reduced to 720 compounds, of which all showed >75% inhibition. A small subset (36 compounds) was selected based on physicochemical properties, known pharmacology, and absence of known ATP-competitor motif, to determine mechanism of action by performing inhibition dose-response curves at eight different ATP concentrations. Nonlinear regression was used to calculated Vmax and Km(app) for ATP at 10 different inhibitor concentrations. Twenty-four of 36 investigated compounds displayed a typical ATP-competitive mechanism of inhibition, as exemplified by compound AZ5168 (
Characterization of Hits Acting as Inhibitors of TrkA and NGF Interaction
As outlined in Figure 3 (encircled in red), 2929 compounds were considered to be inactive in the TrkA kinase assay (<20% inhibition), despite the fact that they were active in the TrkA PathHunter cell-based assay (>40% inhibition). Given the large number of hits that were not active in the TrkA kinase assay, there was a need to identify the molecular mechanism of action for these compounds. One plausible explanation could be that they were false-positives due to interference with readout, likely due to inhibition of β-Gal. Thus, a subset of 25 compounds was characterized with respect to substrate interference using the TrkA PathHunter cell-based assay (data not shown). The selection criteria for these compounds were mainly their potency, which ranged from 10 nM to 40 µM in the TrkA PathHunter cell-based assay. The results demonstrated that compounds interfering with the readout (quencher or inhibitors of β-Gal) could at most account for 10% of the actives (data not shown). Previous screens of our corporate compound library have demonstrated the presence of β-Gal inhibitors, and thus, this is the most likely reason for the high percentage of false-positives. Thus, approximately 2600 of 2926 compounds could act through a molecular mechanism not including kinase inhibition or interference with assay readout.
To identify the molecular targets for these compounds, a set of assays was developed that would address interference of the first steps of NGF signaling. The protein-protein interaction between TrkA and NGF was characterized with CisBio’s HTRF technique, NMR spectroscopy, and SPR using BIACORE. Furthermore, direct interaction of compounds with the extracellular domain of TrkA or with NGF was characterized by SPR, where TrkA or NGF was bound to the sensor surface.
First, an HTRF-based protein-protein interaction assay was developed employing biotinylated-NGF (Bio-NGF) bound to streptavidin labeled with Europium cryptate (SA-K) as one component of the HTRF pair. The other pair was an immune complex of TrkA-Fc chimera and anti-human Fc antibody labeled with XL665. Upon interaction of NGF and TrkA, the two fluorophores come in close proximity, and a fluorescence resonance energy transfer (FRET) reaction can occur. In the same manner, no FRET occurs when test compounds interact with either one of the two components, thereby inhibiting the protein-protein interaction. The assay was optimized with respect to amounts of Bio-NGF, TrkA-Fc, SA-K, and XL665-labeled antibody; incubation time; plate type; and temperature (data not shown). The presence of BSA in the buffer was found to have a strong positive effect on assay performance. The Z′ increased to approximately 0.8 when BSA was included in buffers before dilution of both TrkA and Bio-NGF. Absence of BSA leads to negative Z′ values (
Second, an SPR approach using BIACORE, in which one binding partner was immobilized on the sensor chip, was used to investigate the ability of compounds to inhibit the interaction of TrkA and NGF. The SPR approach used continuous flow to study the interactions between binding partners, thus allowing for kinetic studies. In contrast to this, kinetics could not be studied in the HTRF-based assay because equilibrium between binding partners was reached at the time of analysis, as judged by a very low increment of signal over time (
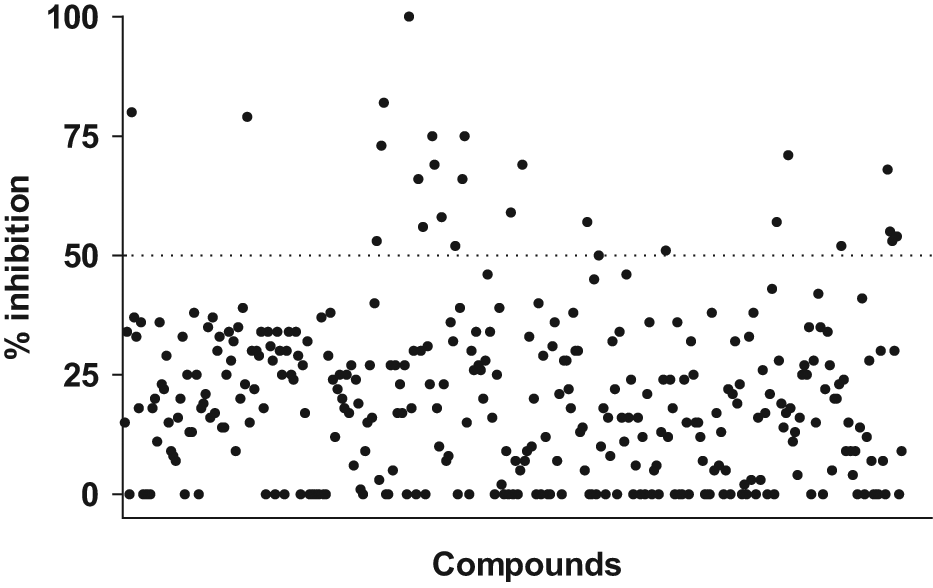
Inhibition of TrkA:NGF interaction using surface plasmon resonance/BIACORE. A total of 70 nM nerve growth factor (NGF) was preincubated with 100 µM test compound and injected to monitor binding of NGF to immobilized TrkA-Fc on the sensor surface. The dotted line indicates compounds inhibiting >50% of the NGF:TrkA binding response. All compounds were measured n = 1.
Third, as a complement to the BIACORE assay described above, we used an NMR displacement assay to demonstrate that compounds could displace a reporter molecule bound to NGF. An internal AstraZeneca compound (AZ3302), which was identified as a reversible binder to NGF in a fragment-based screen (data not shown), was used as a reporter molecule in the NMR displacement assay. In a cross-competition experiment with another small molecule that bound to NGF, the affinity for AZ3302 was determined (Kd = 85 ± 20 µM). Characterization of 1600 compounds was done by the use of the NMR displacement assay, and ligand affinity (Kd) and ligand efficiency were calculated. Eighty compounds demonstrated Kd values less than 10 µM. The correlation of Kd values between NMR and SPR was decent with an r2 value of 0.42 (
Characterization of Hits Acting as Direct Binders of NGF
To characterize the direct binding of small molecules to NGF with a higher throughput than NMR, a direct binding assay was developed in which single-site biotinylated NGF (Bio-NGF) was immobilized to the sensor chip. The Bio-NGF retained is biological activity as judged by the activity in the primary cell-based assay. Bio-NGF had an EC50 value that was slightly higher than wt-NGF, and a fourfold shift in potency was observed (n = 2, data not shown). The final assay conditions, with approximately 3000 RU immobilized NGF, could confirm that the ligand-binding activity for the immobilized NGF was close to 100%. The NGF BIACORE assay was used to characterize 6651 hits. Compound binding was monitored for 60 s followed by a dissociation time of 180 s. Figure 5 shows that 46 of the tested compounds had saturation larger than 50%. Seventeen compounds with Kd values ranging from 0.7 to 100 µM were selected for efficacy studies in the neurite outgrowth assay.
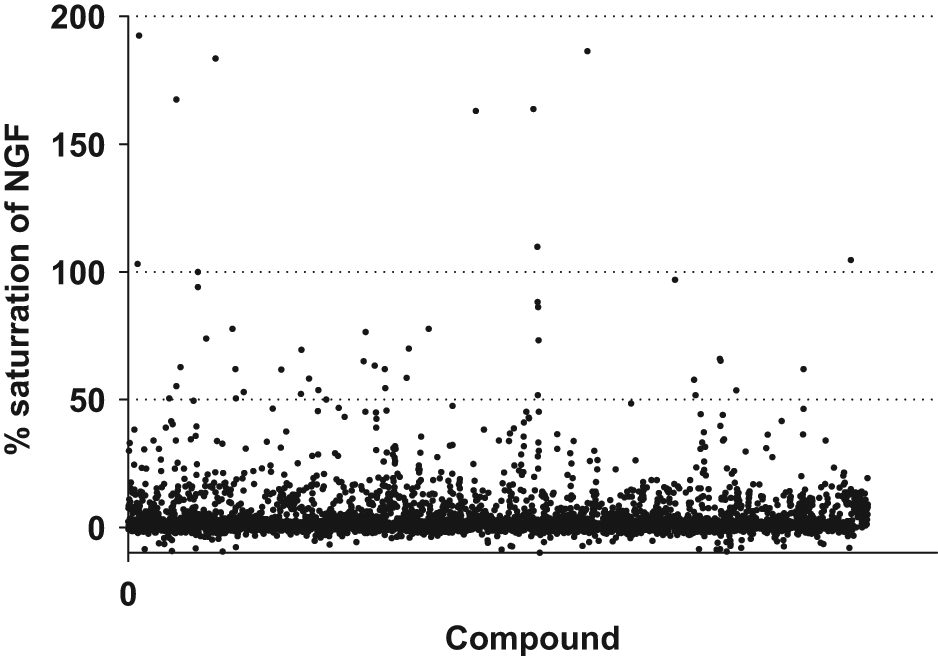
BIACORE response for direct binding of compounds to nerve growth factor (NGF). Test compounds were injected over the sensor chip surface with immobilized Bio-NGF for 1 min at a flow rate of 30 µL/min. Of more than 6500 tested compounds, approximately 400 resulted in a response characterized as active in the direct binding NGF BIACORE assay, but only 46 compounds could saturate the binding to >50%. Each compound was measured n = 4, and compounds were considered as active if the response was greater than three times the standard deviation of a buffer injection.
Efficacy Evaluation of Hits with Neurite Outgrowth Assay
To assess functionally the efficacy of compounds, a neurite outgrowth high-content phenotypic assay was developed using Neuroscreen-1 (NS-1) cells stably expressing green fluorescent protein (NS1-GFP). The GFP-expressing NS-1 cells responded to NGF with increased neurite outgrowth, which was sensitive to TrkA kinase inhibitor (
Effects of Compounds in the TrkA PathHunter Cell-Based Assay, the Neurite Outgrowth Assay (NOG), and the TrkA Kinase Assay.
Discussion
NGF signaling is complex because it can support neurotrophic functions and nociceptive signaling as well as functions on nonneuronal cell populations such as mast cell degranulation. Thus, NGF signaling is dependent on the cellular context. Several previous attempts to identify compounds that intervene with NGF/TrkA signaling have employed biochemical assays.20,24,25 Considering the important role that the cellular milieu have on of NGF/TrkA signaling, a cell-based pathway assay was applied to identify small molecules that could affect NGF/TrkA signaling.
The TrkA PathHunter cell-based assay was demonstrated to be useful in the identification of compounds that affect NGF/TrkA signaling. We have successfully used the assay to screen 677 14;000 compounds. The assay is robust, demonstrates good reproducibility, and is well suited for HTS as well as secondary pharmacology. During this work, we have characterized more than 7200 compounds with respect to IC50 values. Approximately 1% of the investigated compounds displayed IC50 values lower than 100 nM ( Fig. 2C ), suggesting that the assay is well suited for identifying potent compounds in HTS mode. The good correlation between TrkA cells and TrkA/p75 cells ( Fig. 1 ) suggests that the pharmacology, at least for the investigated compounds, is not dependent on the presence of p75.
The HTS hits could partially be ranked based on their cellular activity in the TrkA PathHunter assay. This ranking, in combination with the results from the deconvolution assays, allowed for the selection of compounds for characterization in the neurite outgrowth assay. Although not all investigated compounds were tested in all assays, an assumption was made based on the number of compounds tested in each assay that 57% of the hits were TrkA kinase inhibitors, 22% could inhibit the interaction between TrkA and NGF (based on NGF:TrkA HTRF and NGF:TrkA BIACORE assays), 6% could bind directly to NGF (NMR and NGF BIACORE assays), 10% could interfere with assay readout (promiscuous inhibitors or β-Gal inhibitors), and finally that approximately 5% displayed an unknown mechanism of action. In summary, the identification of compounds with different molecular mechanisms of actions is reported (
The use of the TrkA PathHunter cell-based pathway assay has in this work proven to be invaluable for identifying unique molecular mechanisms of action. This non– target-centric approach needed several secondary assays for hit deconvolution and for identifying the mechanism of action. Given the fact that alternative approaches to intervene with NGF/TrkA signaling were identified, the HTS campaign and the following work were considered very successful.
Footnotes
Acknowledgements
Alf Claesson and Malin Eklund are gratefully acknowledged for supportive discussions, and Ebru Poulsen is acknowledged for support with automation.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.