
Abstract
To begin to develop a high-throughput assay system to evaluate potential small-molecule therapy for Parkinson’s disease (PD), we have performed a low-throughput assay with a small number of compounds using human pluripotent stem cell–derived dopaminergic neurons. We first evaluated the role of 44 compounds known to work in rodent systems in a 1-methyl-4-phenylpyridinium (MPP+) assay in a 96-well format using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay as a readout for neuroprotection. Glial cell–derived neurotrophic factor was used as a positive control because of its well-documented neuroprotective effect on dopaminergic neurons, and two concentrations of each drug were tested. Of 44 compounds screened, 16 showed a neuroprotective effect at one or both dosages tested. A dose-response curve of a subset of the 16 positives was established in the MPP+ model. In addition, we validated neuroprotective effects of these compounds in a rotenone-induced dopaminergic neuronal cell death, another established model for PD. Our human primary dopaminergic neuron-based assays provide a platform for rapid screening and/or validation of potential neuroprotective agents in PD treatment using patient-specific cells and show the importance of using human cells for such assays.
Introduction
Parkinson’s disease (PD) is a debilitating disorder that damages neurons in specific dopaminergic brain areas, causing symptoms such as resting tremor, rigidity, and bradykinesia. Although the symptoms are serious, the onset of these complications is slow. It is estimated that there is a 6-y time frame from the onset of neuronal cell loss and the emergence of clinical dysfunction.1–3 During the past decade, researchers have shown an increased interest in neuroprotection therapy during the early stage of the disease, and several compounds have been evaluated based on known or proposed mechanisms of actions.1–3 Results from such studies have been reviewed in a variety of articles, and well over 150 compounds have been studied in vitro or in vivo. Most of these molecules have been evaluated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-, 6-hydroxydopamine (6-OHDA)-, or the newly described rotenone-based models in rodents, and most have used the catecholamine-synthesizing human cell lines such as SHSY5Y.
Several mechanisms of action have been proposed for these compounds. One class of these molecules is thought to act by modifying dopamine synthesis or reuptake as dopamine itself is considered to be toxic to cells. Other molecules are thought to have a protective effect because of their antioxidant properties. These include antioxidants such as N-acetyl cysteine and resveratrol. Another class of anti-inflammatory drugs includes indomethacin, and this class is thought to modulate the inflammatory cytokine cascade that may contribute to cell death. Others, including minocycline, are thought to modulate mitochondrial bioenergetics, and defects in mitochondrial biology may underlie some familial PD.1–3 Drugs, such as brain-derived neurotrophic factor (BDNF), glial cell–derived neurotrophic factor (GDNF), artemin, persephine, and other members of the GDNF family, may provide trophic support to cells. The mechanism of neuroprotection of drugs such as nicotine is not clear and is based on observations that PD is significantly less prevalent in smokers than in nonsmokers. Given that nicotinic acetylcholine receptor receptors are present in the substantia niagra and that nicotine binds to this class of receptor, it is possible that this binding mediates the activity of nicotine. However, modulation of blood flow may be an equally important role of nicotine.
Standard models have been used for drug screening with nonhuman dopaminergic neurons, or human cell lines, such as SHSY5Y, in vitro, or rodents in vivo. Although these models have limitations, screening with these models has nevertheless allowed the development of some new agents. Azilect (rasagiline), a second generation of monoamine oxidase B (MAOB) inhibitor, was recently introduced into the market, and it appears to be a promising candidate for neuroprotection. Propargylamines, such as rasagiline, have neuroprotective and neurorescuing properties via their effect on the mitochondria, which interferes with and blocks apoptosis in neurodegenerative disorders.4,5 Laboratory studies show that rasagiline has in vitro and in vivo neuroprotective effects, but the mechanism of action underlying its neuroprotective effects in PD patients is unknown at present.
The recent advance in pluripotent stem cell (PSC) technology has allowed us to consider evaluating mechanisms of action of neuroprotective compounds in vitro using normal embryonic stem cell (ESC) lines and induced pluripotent stem cell (iPSC) lines from sporadic PD or from patients with familial PD. Indeed, we and other investigators have shown that it is possible to derive dopaminergic neurons from PSC and maintain them in culture to a stage at which rotenone, 6-OHDA, and MPTP assays can be performed with a variety of readouts. 6 We previously showed that such assays were sensitive even in mixed cultures. 6 Equally important, our results suggested that human sources of cells may be available in sufficient quantities to be able to determine the similarities and differences between species and thereby harvest the rich data sets that are available from previously evaluated compounds.
In this article, we developed a neurotoxic/neuroprotective platform with an automated readout using PSC-derived primary dopaminergic neurons in mixed cultures in 96-well format. We screened a small number of selected compounds known to have efficacy in rodent or human cell line models either in vitro or in vivo that reflect different mechanisms of action proposed to offer neuroprotection. We showed that only 16 of 44 compounds had a neuroprotective effect in the MPTP model and validated our results by retesting a subset in both MPP+ and rotenone models. Our data suggested that PSC-derived primary dopaminergic neurons can be used for screening and/or validation of potential neuroprotective agents for PD.
Materials and Methods
Materials
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 1-methyl-4-phenylpyridinium
Generation of Dopaminergic Neurons from ESC and iPSC
Neural stem cell (NSC) lines derived from ESC (H14) and iPSC (BC1) were cultured in Neurobasal media containing 1× NEAA, 1× L-glutamine (2 mM), 1× B27, and bFGF (20 ng/mL), as previously described. 7 Dopaminergic differentiation of NSCs was induced by medium conditioned on PA6 cells as described previously. 8 Briefly, dopaminergic differentiation in medium conditioned on PA6 cells (PA6-CM) was initiated by culturing NSCs on culture dishes coated with poly-L-ornithine (20 µg/mL) and laminin (10 µg/mL). After 14 days of differentiation, NSCs were transferred to 96- or 4-well plates. Cells were cultured in PA6-CM with BDNF (20 ng/mL) and GDNF (20 ng/mL) for 14 d and then treated with MPP+ or rotenone for 24 h. To evaluate the effect of neuroprotective agents on cell death, these compounds were added 1 h prior to MPP+ or rotenone (Sigma).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde, washed in phosphate-buffered saline (PBS), and then incubated in PBS containing 10% normal goat serum and 0.3% Triton-X for 1 h at room temperature, as described previously. 9 The cells were then incubated with primary antibodies (Sox1, 1:200; Tuj1, 1:1000; GFAP, 1:2000; TH, 1:200; and Nestin, 1:200) in blocking solution overnight at 4 °C. The cells were washed with PBS and incubated with fluorochrome-conjugated secondary antibodies (1:200; Molecular Probes, Inc., Eugene, OR) for 1 h at room temperature. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Control experiments were performed in which one or the other of the primary antisera was omitted. No staining was observed under these conditions. The total number of TH-positive neurons in cultures was counted after 24 h of MPP+ or rotenone treatment. The percentage of TH-positive neurons as compared with that of untreated cells was used to evaluate MPP+ or rotenone toxicity. Experiments were repeated with cultures isolated from independent dissections. 9
MTT Assay
Cell viability was evaluated using an MTT assay, as described previously. 10 Briefly, cells grown in 96-well plates were treated as required. Then, MTT tetrazolium salt (5 mg/mL) was added to each well, and incubation was continued for 2 h at 37 °C. The formazan crystals resulting from mitochondrial enzymatic activity on MTT substrate were solubilized with DMSO. Absorbance was measured at 570 nm using a microplate reader (Molecular Devices, Sunnyvale, CA). Cell survival was expressed as absorbance relative to that of untreated controls.
Gene Expression by Microarray
RNAs isolated from dopaminergic populations were hybridized to Illumina Human HT-12 BeadChip (Illumina, Inc., San Diego, CA, performed by Microarray core facility at the Burnham Institute for Medical Research). Array data processing and analysis were performed using Illumina BeadStudio software. The Illumina array data were normalized by the background method. The maximum expression value for a probe set of one gene was chosen as the expression value of this gene. A differentially expressed gene was defined if the gene showed twofold expression change between any two samples. All cell line correlations were a measure of Pearson’s rho implemented in SAS.
Statistical Analysis
All data are expressed as mean ± SEM for the number (n) of independent experiments performed. Differences among the means for all experiments described were analyzed using one- or two-way analysis of variance. Newman-Keuls post hoc analysis was employed when differences were observed by analysis of variance testing (p < 0.05).
Results
Developing a Screening Platform with Human Primary Dopaminergic Neurons: Culture of Dopaminergic Neurons in 96-Well Format and Validation of Assays for Measuring Toxic and Neuroprotective Effects
We have previously reported dopaminergic-inducing activity of medium conditioned on stromal cell line PA6 (PA6-CM) and shown that exposure to PA6-CM at the NSC stage was sufficient for the acquisition of a dopaminergic fate, in which approximately 25% of cells expressed TH (a dopaminergic neuron marker) after 4 wk of differentiation. 8 To adapt this culture system for screening, we first optimized the differentiation process for 96-well plates, a format that is suitable for low- to medium-throughput screening, using NSC derived from the ESC line H14. NSCs were cultured in PA6-CM for 2 wk and then plated onto 96-well plates for 2 more weeks in the same medium plus BDNF and GDNF. Dopaminergic neuronal differentiation was efficient as quantification of TH-positive neurons after 4 wk of differentiation by immunostaining showed that 30% ± 6% of total cells expressed TH ( Fig. 1A ). We then tested the differentiation process using NSCs derived from an iPSC cell line, BC1, and obtained similar (although slightly lower efficiency, with about 26% ± 4% of total cells expressing TH after 4 wk of differentiation in 96-well plates ( Fig. 1A ). The percentage of differentiation was consistent from well to well and from batch to batch (data not shown). Cell could not, however, be frozen within the 96-well format, and cultures required feeding every 2 d. Thus, the assay setup required a 4-wk culture period prior to initiation of the actual screening experiment.
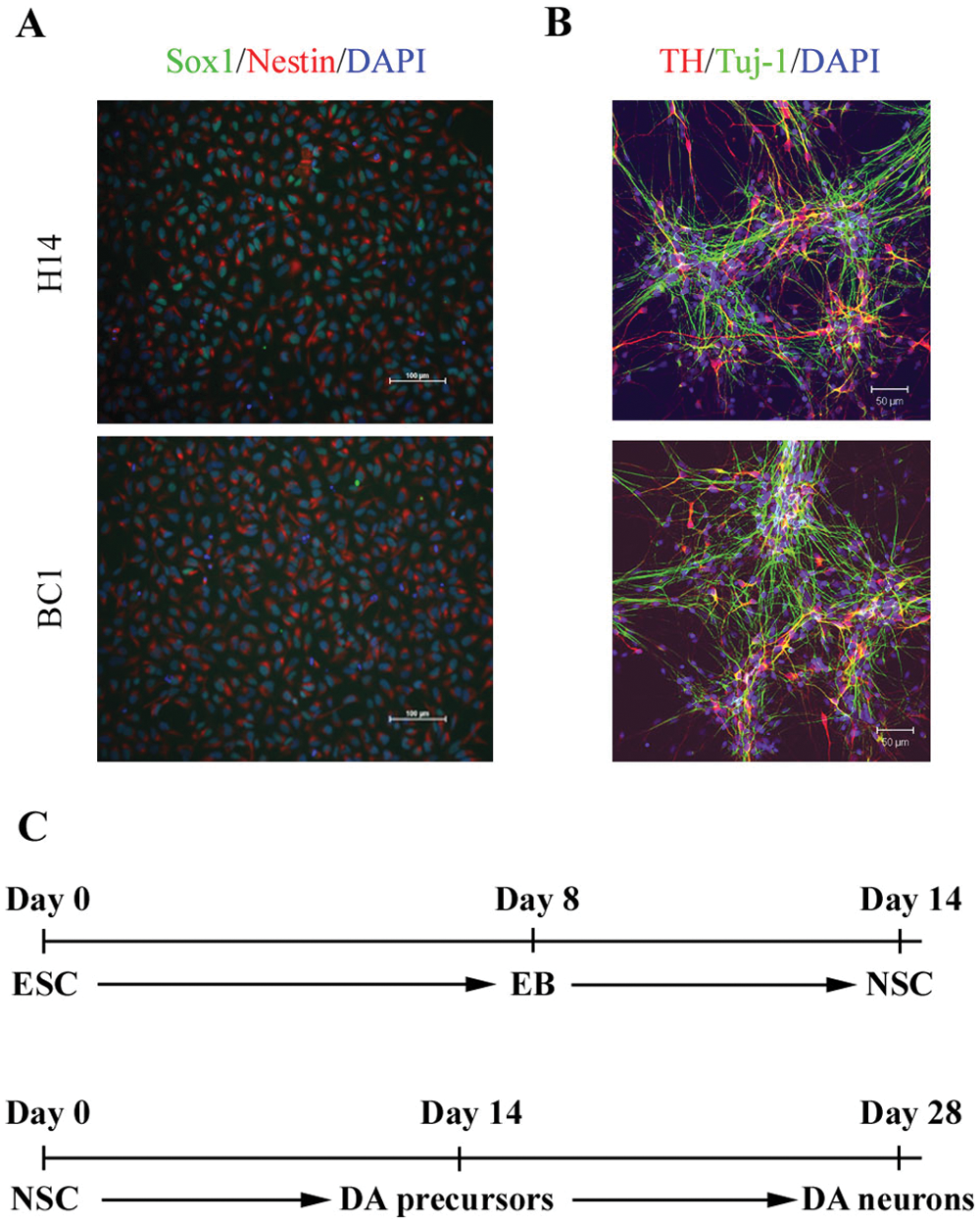
Generation of dopaminergic neurons from embryonic stem cell/induced pluripotent stem cell–derived NSC. (
To confirm the midbrain dopaminergic neuron characteristic of our culture, we examined the whole genome expression profiling of cultures at the assay stage and, in particular, the expression of dopaminergic- and PD-related genes. As seen in
To scale up and to shorten the culture period for experiments, we examined whether cells could be stored at the NSC stage, a stage in neural differentiation of PSCs, and then plated into 96-well plates.7,11 Our laboratory has derived more than 20 NSC lines from ESCs and iPSCs and validated that the NSC lines can be cultured for prolonged periods in vitro without losing their NSC identity or their ability to generate midbrain-specific dopaminergic neurons. For the two NSC lines used in this study, more than 95% of the cells in the NSC cultures were double-labeled with Sox1 and nestin ( Fig. 1A ) with less than 4% β-III tubulin–positive neurons and no GFAP-positive astrocytes nor GalC-positive oligodendrocytes (data not shown).
To further explore the ability to shorten the prescreening time period, we tested whether cells at later stages of dopaminergic differentiation can be cryopreserved without affecting their ability to generate dopaminergic neurons. We found that cells stored at up to day 14 (after the NSC stage) differentiated into dopaminergic neurons at an efficiency similar (measured by the percentage of TH+ neurons by immunostaining) to cells that had not gone through freeze/thaw cycles (data not shown). This significantly reduced the length of culture period for developing and validating assays for screening described below. A flow chart of the differentiation process with time points of cell culture, storage, and assay is shown in Figure 1B . We tested the protocol with multiple ESC- and iPSC-derived NSC lines, including the two lines (H14 and BC1) used in this study, and showed that the protocol and process allowed us to generate sufficient numbers of cells for running assays. 8
To determine which assays of dopaminergic neuronal toxicity and protection as well as automated readouts for such effects would be optimal for this primary cell population in a 96-well format, we tested a number of toxicants, control protective agents, viability, and metabolic activity readouts. 6 For the dopaminergic toxicity model, we found that MPP+, an oxidative metabolite of a well-known dopaminergic toxin, MPTP, 12 worked well. As shown in Figure 2A , a dosage-responsive dopaminergic neuronal cell death measured by the numbers of TH+ neurons by immunostaining was observed after 24 h of exposure to MPP+ in day 28 culture. Approximately 50% of total TH+ neurons remained viable after exposure to 500 µM MPP+ for 24 h. The fact that little effect was seen in the nonneuronal populations at this dose allowed us to selectively assess dopaminergic neuron toxicity in a mixed culture model. For neuroprotection, we chose to test GDNF, a growth factor that has been shown to have a protective effect in dopaminergic neurons both in vitro and in vivo. Figure 2 showed that GDNF reversed MPP+-induced toxicity in dopaminergic neurons to about 50% at the dosage of 20 ng/µL and about 90% when a higher dosage (40 ng/µL) was used.
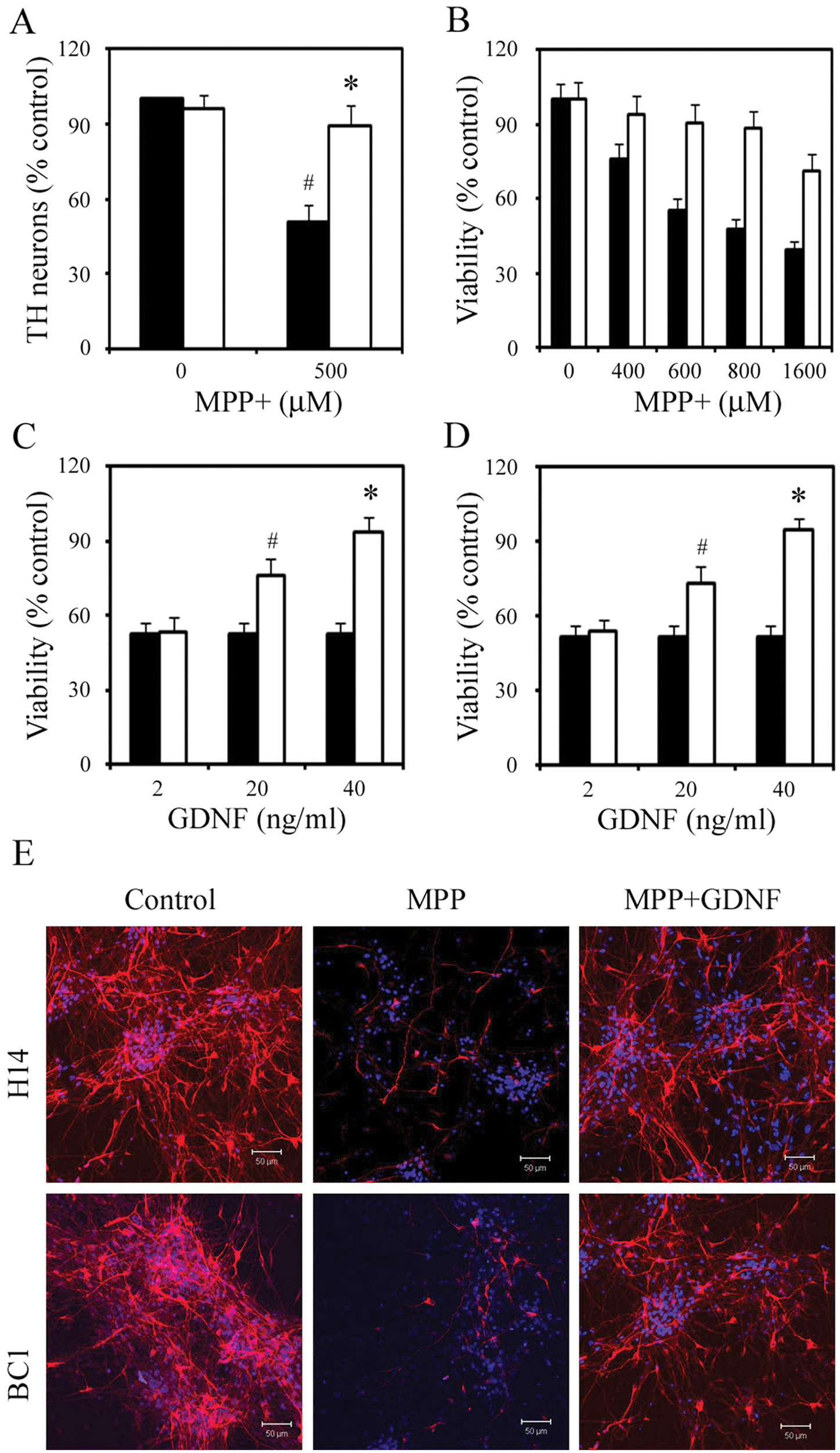
Dopaminergic neurotoxic and protective models. Day 28 cells were treated with glial cell–derived neurotrophic factor (GDNF) 1 h prior to the addition of 1-methyl-4-phenylpyridinium (MPP+). (
We further assessed the validity of the MPP+ toxic and GNDF neuroprotective models in our dopaminergic neuron mixed culture system using the MTT assay as readout, which is suitable for high-throughput screening. As shown in Figure 2 , cell death measured by MTT assay for both MPP+ toxicity and GDNF neuroprotection was consistent with quantification of TH+ neurons measured by immunostaining. About 50% of cells remained viable by MTT assay when exposed to 600 µM MPP+ for 24 h ( Fig. 2B ), and when GDNF was added in the culture, approximately 90% of the cells survived ( Fig. 2C and 2D ).
We also examined the utility of the rotenone model of toxicity for human PSC-derived dopaminergic neurons grown in a 96-well format, and the model worked well using MTT as a readout (see the last subsection of the Results section). We chose to proceed with the MPP+ model as a first-pass method with a secondary validation with the rotenone model using MTT as assay readout. These results suggest that screening for molecules of dopaminergic neuron toxic and protective effects is possible with primary neurons in a platform (96-well format with a readout, MTT assay, that can be automated) suitable for the development of high-throughput screening.
Primary Screen of 44 Compounds
To test the validity of the platform described above for screening for molecules of dopaminergic neuroprotective effect, we obtained 44 compounds that have been reported in the literature as neuroprotective agents (
Only 16 of the 44 compounds showed a neuroprotective effect against MPP+-mediated cell death at one or both dosages tested ( Fig. 3 ), and many of these compounds were as neuroprotective as GDNF, which is currently in clinical trials. 13 Among them were several known human dopaminergic neuron protective agents, such as selegline, nicotine, and a variety of antioxidants ( Fig. 3 ). Based on the mechanisms of action proposed in the literature, we categorized the positive compounds into several groups including antioxidant, antiinflammatory agent, MAO inhibitor, and other neurotransmitter ( Table 1 ).

Drug screening. Cell viability in H14 cultures was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) absorbance and expressed as a percentage of viability of untreated control. (
Positive Compounds Categorized by Mechanisms of Action.
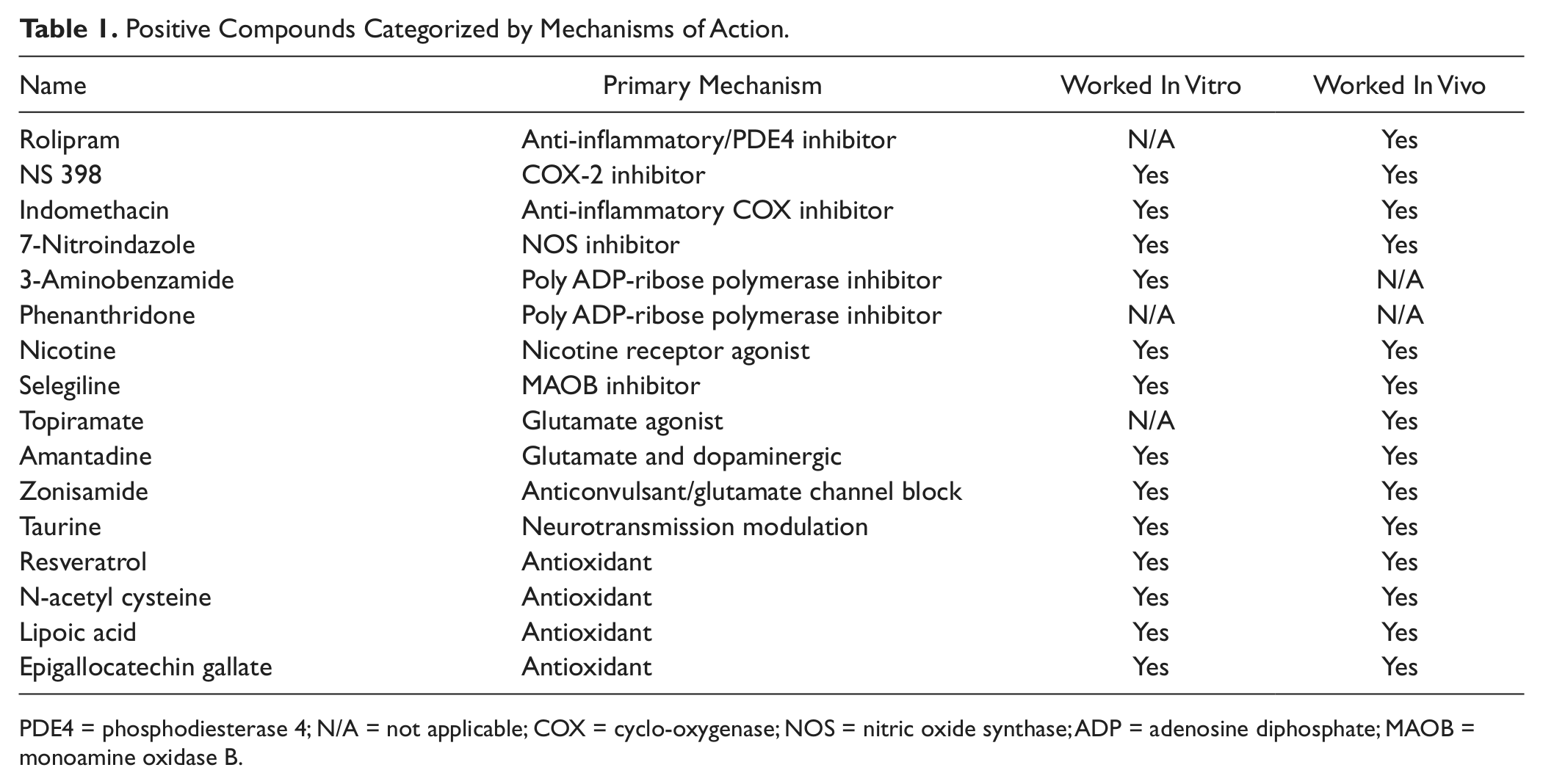
PDE4 = phosphodiesterase 4; N/A = not applicable; COX = cyclo-oxygenase; NOS = nitric oxide synthase; ADP = adenosine diphosphate; MAOB = monoamine oxidase B.
We noted that several compounds, including rasagiline and creatine, which are currently used in clinical trials for PD, did not show a positive effect in our primary screening, which could be due to an inappropriate dose used (we tested only two doses of all compounds) or the differences between species (mouse and human) or between primary cells and immortalized cell lines. Nevertheless, our results indicate that our screening system is a rapid predictor of potential disease-modifying agents for PD. The use of 96-well format and the MTT assay made it feasible to be adapted to a high-throughput screening system using human primary dopaminergic neurons.
Retest of Selected Compounds in the MPP+ Model and in an Additional Line
To test the validity of the initial screen, we performed validation tests. We first chose 3 compounds from the 16 compounds that showed a neuroprotective effect in the primary screen and retested their neuroprotective effect using multiple dosages in the MPP+ model with H14-derived dopaminergic neurons. We selected one compound from each of the following categories: antioxidant, antiinflammatory agent, and the MAO inhibitor, and 3 dosages of each compound that likely have a neuroprotective effect based on literature search. As shown in Figure 3 , treatment with 600 µM MPP+ for 24 h caused about 50% cell death in the day 28 cultures. However, when nicotine, indomethacin, and epigallocatechin gallate were added in the culture 1 h prior to the addition of MPP+, cell death was significantly inhibited. In the case of 40 µM nicotine, 140 µM indomethacin, and 40 µM epigallocatechin gallate, MPP+-mediated cell death was inhibited in dopaminergic neuron culture by 57%, 53%, and 49%, respectively ( Fig. 3 ). Neither nicotine, indomethacin, nor epigallocatechin gallate at the concentrations used was stimulatory or inhibitory for neuronal survival in both H14 and BC1 cultures without MPP+ (data not shown).
We realized that our primary screening might miss positive compounds because of the limited dosages used (two dosages only). By carefully examining the literature, we noted that among the negative compounds, the reported dosages of neuroprotective effects by creatine and rasagiline were outside the range chosen in our primary screen. In the case of rasagiline, only doses lower than 10 µM were reported to be neuroprotective, and in the case of creatine, only much higher doses (higher than 100 µM) were shown to be neuroprotective, suggesting that our failure was not due to the sensitivity of the assay. We therefore retested these two compounds at three more appropriate dosages for creatine (2, 20, and 40 mM) and rasagiline (1, 100, and 1000 nM), and the results are shown in Figure 4 . Two doses of each creatine (20 and 40 mM) and rasagiline (100 and 1000 nM) showed a neuroprotective effect in the MPP+ model: MPP+-mediated cell death was inhibited by 82% for creatine (40 mM) and 80% for rasagiline (1000 nM; Fig. 4). These results were further confirmed in the BC1 iPSC line, in which 40 mM creatine and 1 mM rasagiline inhibited MPP+-mediated cell death by 81% and 84%, respectively ( Fig. 4 ). Neither creatine nor rasagiline at the concentrations used were stimulatory or inhibitory for neuronal survival in both H14 and BC1 cultures without MPP+ (data not shown). Finally, the neuroprotective effects measured by the MTT assay were independently validated by immunofluorescence with TH. Cells were stained for TH at 24 h after MPP+ treatment with or without compounds, and TH-positive cells were counted. As shown in Figure 4 , treatment with creatine or rasagiline protected TH-positive neurons from MPP+-induced death.
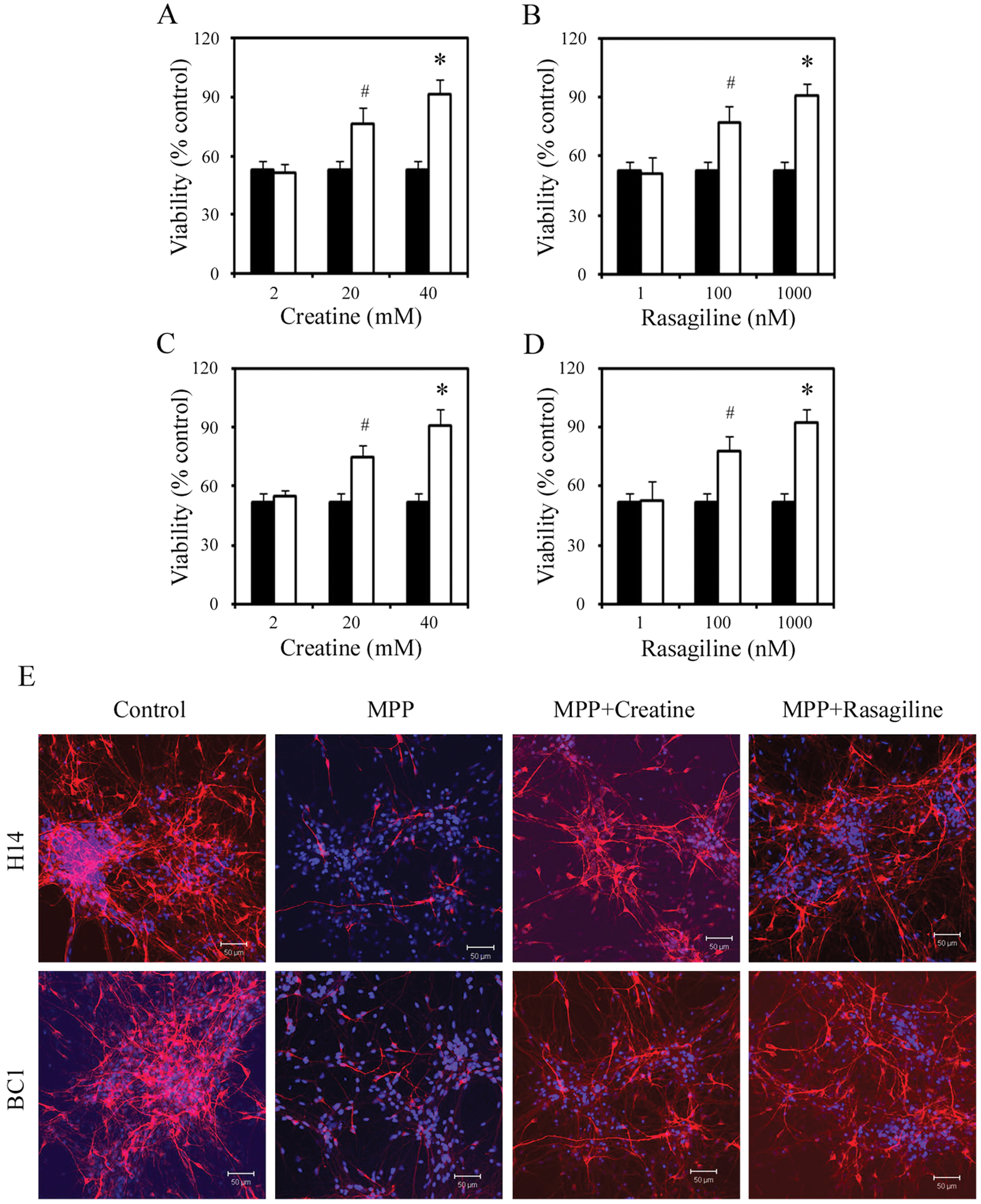
Secondary screens: retesting of negative compounds. Cell viability in H14 (
Validation of Neuroprotective Effects of Selected Compounds in an Additional Toxicity Model
We wished to test/validate some of our screen results in an additional toxicity model and chose to use rotenone, a pesticide that causes highly selective nigrostriatal dopaminergic degeneration that is associated behaviorally with hypokinesia and rigidity. 14 We first determined the optimal dose of rotenone for the assay by performing a dose-responsive analysis in both H14- and BC1-derived dopaminergic neurons. After 4 wk of differentiation, H14- or BC1-derived dopaminergic neurons in 96-well plates were treated with rotenone for 24 h, and cell viability was determined by MTT incorporation. 10 As shown in Figure 5 , treatment with 800 nM of rotenone resulted in about 50% cell death in both H14 and BC1 cultures. We then validated this result by TH staining and counting ( Fig. 5 ). Based on these results, 800 nM of rotenone was used for the neuroprotective experiment described below.
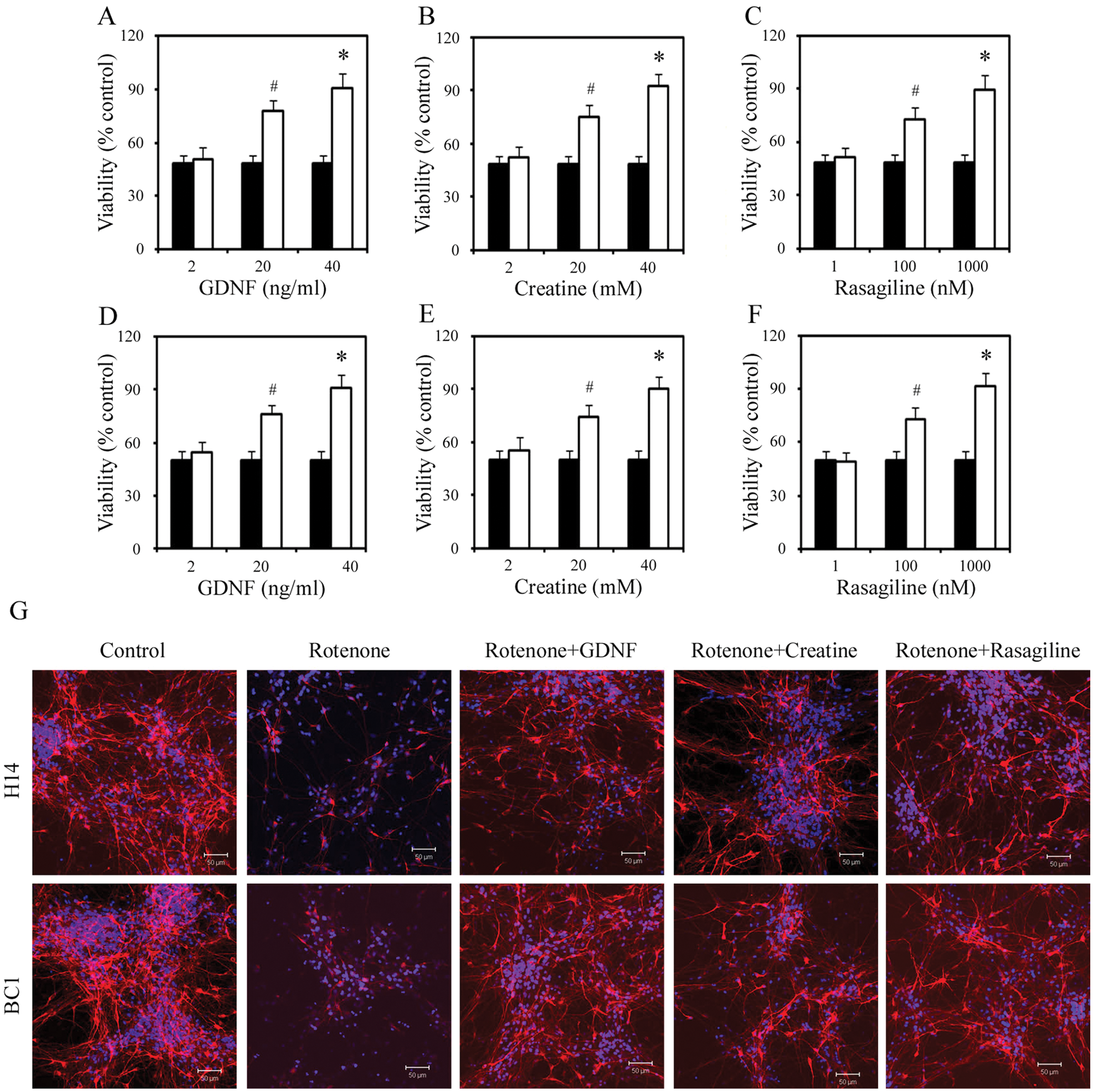
Protective effects of glial cell–derived neurotrophic factor (GDNF), creatine, and rasagiline on rotenone-mediated dopaminergic neuronal death. Cell viability in H14 (
Similar to the MPP model, we tested the effect of GDNF, creatine, and rasagiline on rotenone-induced cell death 1 h before the addition of the toxin and assayed the cell death by MTT 24 h in both H14 and BC1 cultures. In H14 culture, MPP+-mediated cell death was inhibited by 40 ng/mL GDNF, 40 mM creatine, and 1 mM rasagiline by 82%, 86%, and 80%, respectively ( Fig. 5 ). In BC1 culture, 40 ng/mL GDNF, 40 mM creatine, and 1 mM rasagiline inhibited rotenone-mediated cell death by 81%, 80%, and 83%, respectively ( Fig. 5 ). These neuroprotective results were further validated by quantification of TH neurons using immunostaining ( Fig. 5 ). No changes in cell survival/death were observed when GDNF, creatine, or rasagiline at the concentrations used in the cultures when rotenone was not present (data not shown).
Discussion
In vitro cell-based models provide important tools for drug discovery. Tumor-derived or genetically transformed cell lines have been the traditional platforms for screening candidate neuroprotective agents in PD treatment. In this study, we evaluated the utility of PSC-derived populations for screening. In pursuing this goal, we established a screening platform with assays suitable for automated readout using primary dopaminergic neurons derived from ESCs/iPSCs and ran a small screen of compounds for neuroprotective effects in toxic models. Our results show that it is possible to run small screens with primary cells derived from ESCs and iPSCs and obtain consistent and reliable results that offer advantages over screens run in cell lines or in rodent cells. Our results highlight the advantages and limitations of such primary cell–based screens. Although it is difficult to run high-throughput screens, our assays can certainly be used to study the mechanism of action and possible synergic effects of combination of drugs. In addition, these assays allow for evaluation of long-term effects of exposure to a particular treatment/drug because cells can be maintained in culture for prolonged periods.
Although we have previously published on defined medium conditions and defined factors to generate dopaminergic neurons7,11 and have developed sorting protocols to enrich for dopaminergic neurons, we chose to use mixed cultures and an undefined medium condition rather than purified cells or specific factors, as our goal was to understand how these drugs would work in vivo. We selected 44 compounds from the literature, which have been shown to have neuroprotective effects in MPP+, 6-OHDA, or the rotenone model using nonprimary cells of human origin. Almost all of them have also been reported to have efficacy in rodent models in vivo. Nevertheless, only one third of them were positive in our assay. This may reflect that the assay is not adequate or may be because the earlier assays are not as reflective of the human disease. We think the latter is the more likely case because the positive compounds the assay identified are mostly those that have been used in human trials, including two MAO inhibitors that have specific neuroprotective effects, rasagiline and selegline, as well as identifying indomethacin and nicotine, whereas several compounds that have failed clinical trials showed no effect in our assay. Moreover, the lack of efficacy of the other compounds cannot be attributed to the more limited sensitivity of the assay, the stage of maturation of the cells, or the novelty of the assay as we carefully tested multiple parameters in developing the assay. We carefully assessed the gene expression profile of the cells and determined that the correct sets of target genes were being expressed and chose a stage of maturation in which a dynamic range of MPP+ response was present. This development time took 4 wk after NSC maturation. Importantly, we also showed that this process could be performed in a 96-well plate. Cells at this stage expressed many major genes known to cause familial PD, and the cultures did not contain any contaminating macrophages. At this stage, the cultures did not contain any significant number of astrocytes but contained other neurons and stem and precursor cells.
We used GDNF, a member of the transforming growth factor-β superfamily, as a positive control in our assay based on previous results. 6 In vitro, there are numerous studies that have demonstrated that GDNF is a survival factor for primary mesencephalic dopaminergic neurons and protects them against a number of toxic insults.15,16 In vivo, GDNF has been shown to enhance dopaminergic neuron function in 6-OHDA–lesioned rats 17 and protect against MPTP toxicity in mice 18 and monkeys. 19 The compounds that were positive in our screens worked at least as well as GDNF in our assay. In addition, previous results have suggested that these compounds cross the blood-brain barrier to achieve therapeutic concentrations, suggesting the possibility that an in vitro assay may help assess analogues of these compounds, assess the synergy of these compounds, and develop better acting compounds using this in vitro assay.
Our results show that rasagiline and selegline but not other MAOB inhibitors had a protective effect in both rotenone- and MPP+-based assays. Given the absence of astrocytes in our culture and the failure of other MAOB inhibitors to show the same neuroprotective effect, we believe that these may act by a mechanism other than MAOB inhibition as is consistent with other published reports.4,5 Neuroprotection by rasagiline and selegline appears to be related to a propargyl ring incorporated within the rasagiline molecule rather than to MAOB inhibition. Rasagiline increases the expression of the anti-apoptotic B-cell lymphoma-2 (Bcl-2) family proteins via the protein kinase C pathway and reduces the proapoptotic Bcl-2–associated death promoter (Bad) and Bcl-2–associated X protein (BAX). 4 Rasagiline also increases the expression of the neurotrophins BDNF and GDNF. 5 Importantly, rasagiline reduces the long-term progression and symptoms in Parkinsonian patients. 20 Our assay model will allow us to further probe the mechanism of action of these molecules.
A subset of the molecules reported to have antioxidant effects in either rodent models or in SHSY5Y cells were effective in our model system ( Table 1 ). Mitochondria are thought to produce more than 90% of the cellular adenosine triphosphate in neurons, which supports many neuronal functions. There is strong evidence that mitochondrial impairment plays a role in the pathogenesis of PD. Both MPP+21 and rotenone 14 were found to be mitochondrial poisons that inhibit cellular respiration through the blockade of the electron transport enzyme complex I (NADH:ubiquinone oxidoreductase). Several laboratories have reported that there is a selective defect in complex I of the mitochondrial electron transport chain in the substantia nigra of postmortem tissue of PD patients, and there is also reduction of complex I activity in platelets of patients with early PD.22,23
Nicotine, which is currently used in clinical trials in treating PD, is one of the positive compounds in our screen. Several anti-inflammatory agents such as indomethacin and rolipram showed a protective effect in our screen as well. We note that it is likely that each of these molecules acts via a distinct mechanism, and synergy is likely. Our novel model will allow us to test this directly in cells of the appropriate phenotype over the longer time period that is required to assess these interactions. Finally, we note that rasagline and creatine are protective in two different modes of toxic insult and thus are likely to work with familial PD, although direct experiments will be required to validate this.
In summary, we show that dopaminergic neurons derived from ESC and iPSC lines are a promising cell source for screening for drug targets that interrupt this disease processes. We describe a method using differentiated dopaminergic neurons derived from H14 and BC1 NSCs that permits the rapid screening of potential neuroprotective agents in PD treatment. These novel in vitro models might also be helpful in studies of various cellular and molecular pathways that are involved in the normal function and degeneration of human dopaminergic neurons. The use of this tool in high-throughput screening assays should allow better prediction of the therapeutic responses induced by newly developed neuroprotective agents and offer insight into the underlying mechanisms. Given the ability to make dopaminergic neurons from normal, familial, and sporadic PD patients, we believe that using such an assay as a first step to assess purported disease-modifying agents will reduce the time taken for such agents to reach the clinic, as well as increase the success of clinical trials or the possibility of personalizing therapy. We predict that the most effective agents will be those that protect against the multiple cellular dysfunctions linked with PD.
Footnotes
Acknowledgements
The authors thank Drs Wei Zheng and Manju Swaroop and the compound management team at the National Center for Advancing Translational Sciences for providing the compounds.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by California Institute for Regenerative Medicine Grants TR-01856 and CL1-00501 to X.Z. and the NIH CRM, a common fund initiative, to M.S.R. The authors thank Drs. Wei Zheng and Manju Swaroop and the compound management team at the National Center for Advancing Translational Sciences for proving the compounds.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.