
Abstract
TRPV1 was originally characterized as an integrator of various noxious stimuli such as capsaicin, heat, and protons. TRPV1-null mice exhibit a deficiency in sensing noxious heat stimuli, suggesting that TRPV1 is one of the main heat sensors on nociceptive primary afferent neurons and a candidate target for heat hypersensitivity in chronic pain. Several different potent and selective TRPV1 antagonists have been developed by more than 50 companies since the characterization of the receptor in 1997. A consequence of this competitive interest is the crowding of patentable chemical space, because very similar in vitro screening assays are used. To circumvent this issue and to expand our understanding of TRPV1 biology, we sought to take advantage of recent advancements in automated patch-clamp technology to design a novel screening cascade. This SAR-driving assay identified novel modulators that blocked the depolarization-induced activation of outwardly-rectifying TRPV1 currents independent of agonist stimulation, and we correlated the pharmacology to three other innovative assays for higher-throughput screening. Ultimately, we have identified a screening paradigm that would have good predictive value for future TRPV1 drug discovery projects and novel chemical space with a higher probability of gaining intellectual property coverage.
Introduction
Capsaicin, the active ingredient in chili peppers, has long been known to cause nocifensive behaviors in both rodent and man and has been proposed as a target for the treatment of chronic inflammatory pain.1,2 Its receptor, TRPV1, was first cloned by Caterina et al. 3 in 1997, initiating a new wave of pharmacologic research to harness the therapeutic potential from this nonselective cation channel. Both agonists to desensitize nociceptive small-diameter afferent neurons, and antagonists to block pain-evoked neuronal transmission, have been discovered for this channel. 4 However, more monetary and intellectual investment has been placed on the latter type of therapeutic, with more than 50 different pharmaceutical and biotechnology companies reporting research and development activities. 5 A consequence of this popularity within the pharmaceutical industry is the extensive amount of patentable space that is covered. At last count, 511 patents that cite the discovery of small- and large-molecule TRPV1 modulators have been filed (Scifinder, American Chemistry Society and Voight and Kort 6 ). Most of these compounds are extremely lipophilic, like capsaicin itself, and have very poor aqueous solubility that has been problematic for the development of these compounds in clinical trials. For the most part, the discovery of small-molecule TRPV1 antagonists has relied on high-throughput screening (HTS) technology that measures differences in intracellular calcium concentrations. Many of the early lead compounds, including BCTC, 7 SB-366791, 8 and AMG9810, 9 as well as clinical candidates, such as ABT-102, 10 AMG-517, 11 and SB-705498, 12 were identified using fluorescent imaging plate reader (FLIPR) Ca2+ influx or 45Ca2+-uptake assays. These assay methods measure the ability of chemical entities to block an agonist-induced Ca2+ flux through the TRPV1 channel, with either capsaicin, protons, or heat (>42 °C) used to activate the channel. 3 In most of the reported and patented cases, however, capsaicin is used as the preferred ligand to probe TRPV1 SAR under HTS conditions.
When TRPV1 was first cloned, it was described not only as a ligand-gated channel that responded to capsaicin and protons but also as a weakly voltage-gated channel with outward rectification properties. 3 In fact, this is a trait shared by many other members of the TRP channel family. 13 The recent advancements in the area of automated patch-clamp technologies have opened the door to higher-throughput pharmacological screening for many voltage- and ligand-gated ion channels.14–20 To explore the potential of a new chemical landscape for TRPV1, we took advantage of the voltage-gated properties of TRPV1 and developed a medium-throughput automated patch-clamp assay method using the IonWorks Quattro system. Although this method proved to be very effective in identifying novel chemical entities for TRPV1, its potential for use in an HTS screening paradigm was limited in terms of the cost per data point and throughput capabilities. Three other novel TRPV1 screening platforms were validated, namely, (1) the voltage-sensitive dye detection of membrane potential variation after proton-gated stimulation of TRPV1, (2) atomic absorption spectroscopic measurement of spontaneous rubidium-flux through TRPV1, and (3) the Ca2+-induced activation and Ca2+-influx of TRPV1. By correlating the quantity and quality of the hits generated from each of these three novel assays with the automated electrophysiological assay, we identified the atomic absorption method as an initial HTS prior to using a lower-throughput but data-rich electrophysiology method. The development and validation of these assays are described here, along with the correlations that enabled the implementation of an innovative screening cascade to discover new TRPV1 antagonists.
Methods
Stably Expressing Inducible TRPV1 Cell Lines
Human TRPV1 sequence (accession No. AX002940) was subcloned in a pcDNA4/TO inducible expression vector (Invitrogen, Carlsbad, CA) and then transfected into Chinese hamster ovary (CHO) or human embryonic kidney (HEK) cells stably expressing the T-REx repressor gene (Invitrogen) using an electroporation method as previously described. 21 A stable pool was generated by maintaining the cells in DMEM/F12 culture medium (Wisent, St-Bruno, QC, Canada) supplemented with 10% fetal bovine serum, 1% glutamine, and 350 µg/mL Zeocin and 5 µg/mL Blasticidin (Invitrogen). Colonies were selected and plated in a 96-well plate at a concentration of 1 cell/well and maintained in the presence of the selection antibiotics. Clones that survived the selection process were assessed for the functional expression of TRPV1 using the standard capsaicin Fluo-4 FLIPR experimental protocol described below. The highest expressing clones were further characterized for expression stability over a span of 3 mo. The highest stable clone was used for all subsequent assays.
Standard Capsaicin FLIPR Assay with Fluo-4 Ca2+ Detection
hTRPV1-HEK cells were plated in a black, clear-bottom 384-well plate coated with poly-D-lysine (Becton Dickinson Labware, Bedford, MA) at a concentration of 10 000 cells per well in 50 µL and incubated overnight at 37 °C and 5% CO2 along with 0.1 µg/mL tetracycline to induce the expression of TRPV1. The following day, cells were washed in an assay buffer of 1xHBSS (without Ca2+ or Mg2+; Wisent) supplemented with 5 mM glucose and 1 mM CaCl2 before being loaded with 2 µM Fluo-4 AM (Invitrogen) for 1 h. The cells were washed three times before adding 25 µL of assay buffer. The plates were read in a FLIPR-384 instrument during the application of 12.5 µL fourfold the final concentration of test compound prepared in a separate 384-well plate in serial threefold dilutions, and then the subsequent addition of 20 nM capsaicin (final, prepared as fourfold final concentration stock of 80 nM in a 384-well compound plate). Responses to both test compound and capsaicin additions were recorded at 1 Hz for 60 s to capture the acute effects and then at 0.1 Hz for an additional 280 s. Data were extracted and analyzed using FLIPR-384 software (Molecular Devices, Sunnyvale, CA) and Microsoft Excel.
IonWorks Quattro Voltage-Ramp Assay
hTRPV1-CHO cells were maintained as described above. On the day prior to the experiment, cells were induced with 0.1 µg/mL tetracycline. Cells were washed and prepared for the experiment as per the manufacturer’s instructions. The final cell pellet was resuspended to a concentration of 1.2 × 106 cells/mL in Dulbecco’s phosphate-buffered saline (D-PBS; with Ca2+ and Mg2+ (Wisent). For the experiment, D-PBS (in mM NaCl 136.9, KCl 2.7, KH2PO4 1.5, Na2HPO4 8, CaCl2-2H2O 0.9, and MgCl2-6H2O 0.5; Wisent) with 10 mM glucose was used as the extracellular assay buffer. The intracellular solution was composed of the following (in mM): K-gluconate 100, KCl 40, MgCl2 3.2, HEPES 5, and EGTA 3, pH adjusted to 7.2 with KOH. An additional buffer with 0.1 mg/mL amphotericin B was prepared to gain access to the cells via the perforated patch method using the IonWorks instrument.
Cells were loaded onto the instrument, and once a seal was formed, perforated access was achieved with the amphotericin B solution. Cells were maintained at a holding potential of −60 mV, and in order to elicit TRPV1 voltage-gated currents, continuous voltage-ramp sweeps of 500 ms from −80 mV to +80 mV at 1 Hz were applied to the cells prior (at baseline) and after compound addition. The peak amplitude for the resulting outward current at +80 mV was used to compare compound effects on TRPV1.
FLIPR Assay with Voltage-Sensitive Reporting Dye and pH Stimulus
hTRPV1-HEK cells were prepared, plated, and induced in the same manner as described above for the standard FLIPR assay with capsaicin. The day of the experiment, culture media were removed by inversion and then the cells were loaded with the blue membrane potential dye (Molecular Devices) prepared in 1x-HBSS (with Ca2+ and Mg2+, Wisent) for 1 h. After the incubation with the membrane potential dye, plates were placed directly in the FLIPR 384 instrument and read during the application of 12.5 µL of a pH 5.2 DPBS solution, and the subsequent second addition of a fourfold the final concentration of compound prepared in a separate 384-well plate in serial threefold dilutions. Responses to both test pH and compound additions were recorded at 1 Hz for 60 s to capture the acute effects and then at 0.1 Hz for an additional 280 s. Data were extracted and analyzed using FLIPR-384 software and Microsoft Excel.
Calcium-Induced Activation of TRPV1 Assay
hTRPV1-HEK cells were prepared, plated, and induced in the same manner as described above for the standard FLIPR assay with capsaicin. The day of the experiment, culture medium was removed from the cells by inversion, and the cells were loaded with a Fluo-4 no-wash dye (Fluo-4NW; Invitrogen). Briefly, one vial of the Fluo-4NW kit was diluted in 10 mL of an assay buffer with reduced calcium (in mM): KCl 5.3, KH2PO4 0.4, NaCl 137.9, Na2HPO4 0.34, EGTA 5, D-glucose 10, HEPES 10, NaHCO3 4.5, CaCl2 0.000016. The minimal amount of calcium is required so that the cells remain adherent to the plates. Cells were incubated with the dye for 1 h before reading in a FLIPR-384 instrument during the application of 12.5 µL fourfold the final concentration of test compound prepared in a separate 384-well plate with serial threefold dilutions and then the subsequent addition of an assay buffer containing 4.8 mM CaCl2. Responses to both test compound and high extracellular Ca2+ additions were recorded at 1 Hz for 60 s to capture the acute effects and then at 0.1 Hz for an additional 280 s. Data were extracted and analyzed using FLIPR-384 software and Microsoft Excel.
Atomic Absorption Spectroscopy-Based Rubidium Flux Assay
hTRPV1-CHO cells were maintained as described above. Fifty-six hours prior to the experiment, cells were detached and replated on a black poly-D-lysine–coated 384-well plate (Becton-Dickenson) at a concentration of 10 000 cells/well. Twenty hours prior to the experiment, the cells were induced with 0.1 µg/mL tetracycline. On the day of the experiment, compound plates were prepared at a starting concentration of 40 µM in threefold dilutions, representing four times the final concentration intended for the assay. The induction media were removed, and cells were washed three times with a buffer consisting of (in mM) NaCl 150, HEPES 25, CaCl2 2, MgCl2 1, and Na2HPO4 0.8. Of the wash buffer, 12.5 µL was left in each well after the final wash step, and then 12.5 µL of the fourfold final concentration of test compound or control solutions was added for 10 min at room temperature. Subsequently, 25 µL of 2× rubidium loading buffer (in mM: NaCl 130, HEPES 25, CaCl2 2, MgCl2 1, Na2HPO4 0.8, glucose 10, RbCl 11) was added for 3 h at 37 °C. After loading, the plates were washed eight times in a buffer consisting of (in mM) NaCl 150, KCl 5.4, HEPES 25, CaCl2 2, MgCl2 1, and Na2HPO4 0.8, leaving 12.5 µL per well after the final wash. Plates were frozen overnight and then thawed the next day, and pellets lysed with 80 µL of 1% Triton X-100 solution in water. The ICR8000 single-channel atomic absorption flame spectrometer (Aurora Biosciences, Vancouver, Canada) was used to determine the concentration of rubidium present in cells. The instrument was calibrated by generating a standard curve with known rubidium concentrations prior to each run. Fifty microliters of each well was injected, followed by 200 µL of ICR wash buffer, resulting in the concentration of rubidium as calculated from rubidium’s absorbance of light at 780 nm.
Whole-Cell Patch-Clamp Recordings
hTRPV1-CHO cells were maintained as per the cell culture methods described above. On the day of the experiment, cells were treated with 2 mL Accutase for 2 min before washing with 1 mL 1x-D-PBS. The cells were tapped gently to dissociate them from the culture flask and then placed in an Eppendorf tube. A droplet of cells was transferred to a recording chamber fixed to the stage of an inverted microscope (Olympus) and allowed to settle for 2 min prior to being constantly perfused (0.5 mL/min) with D-PBS, a standard extracellular bathing solution containing (in mM) 137 NaCl, 8.1 Na2HPO4•7H2O, 2.68 KCl, 1.47 KH2PO4, 0.9 CaCl2, 0.5 MgCl2•6H2O, 5.6 glucose, and 0.33 Na-pyruvate at pH 7 and 275 to 290 mOsm. Manual whole-cell patch-clamp recordings were performed using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Patch electrodes were pulled from borosilicate glass (World Precision Instruments Inc., Sarasota, FL) on a Flaming/Brown puller (Sutter Instrument Co, Novato, CA) to form a tip diameter of 1.5 to 2.5 mM, which translated to a 2 to 5 MΩ tip resistance. These electrodes are filled with intracellular pipette solution (pH 7.3, 290 mosM) containing the following (in mM): 8 NaCl, 20 KCl, 1 MgCl2, 10 HEPES, 110 K-glutamate, 10 EGTA, and 4 ATP-Mg.
A small patch of the cell membrane underneath the tip of the pipette is aspirated to form a tight, high-resistance seal between the membrane and the pipette, known as a gigaseal. Negative pressure is then applied to rupture the membrane, thereby establishing a whole-cell configuration. The adjustment of capacitance compensation and series resistance compensation (<15 MΩ) is done prior to recording membrane currents. The currents are recorded at a holding potential of −60 mV. Data acquisition is done with Clampex 9.2 software and stored for offline analysis.
High temperature to activate hTRPV1 is delivered to the individual cells via a DAD-VC Voltage Command and Valve Control System (ALA Scientific Instruments, Farmingdale, NY) coupled to a TC2BIP 2 channel bipolar temperature controller and MPRE8 direct perfusion preheater (Cell MicroControls, Norfolk, VA), which use a computer-controlled micromanifold to quickly release and heat a sample of extracellular solution via an electronic valve. Heated solution is applied for a short 1 s burst, and activation of the hTRPV1 channel is observed as a fast-activating and desensitizing inward current (mostly Ca2+ and Na+ ions). Capsaicin (1 µM) or low-pH 5.2 solutions were delivered to the cells with the same DAD-VC valve control system in 1 s bursts.
Compound Sets
Two sets of compounds were used for comparisons. A small set of 96 compounds was used to first test on the IonWorks voltage ramp assay and determine if it would be sensitive enough for further pharmacologic studies. To compare the higher-throughput capabilities and results of other novel screening methods, a diversity set of 7509 compounds was tested on the voltage-sensitive dye, calcium add-back, capsaicin, and atomic absorption assays. The physical chemical properties of some of the more novel hits identified during this preliminary screening process are presented in the results section in Table 1 . All compounds were synthesized by AstraZeneca plc.
Characteristics of TRPV1 Antagonists Identified by Novel Screening Paradigms.
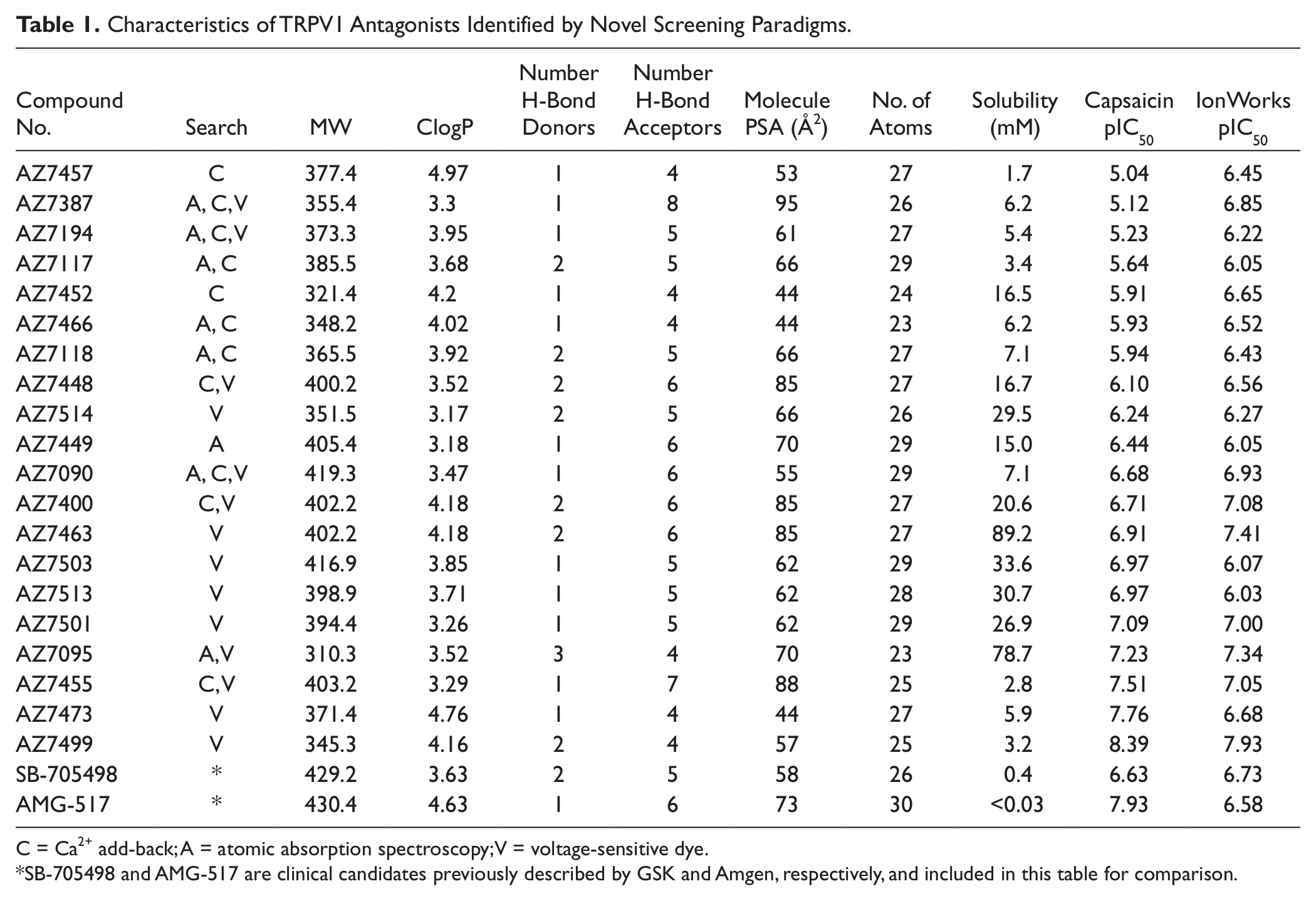
C = Ca2+ add-back; A = atomic absorption spectroscopy; V = voltage-sensitive dye.
SB-705498 and AMG-517 are clinical candidates previously described by GSK and Amgen, respectively, and included in this table for comparison.
Data Analysis and Statistics
Concentration-response curves were generated by fitting data to a four-parameter logistic equation using Prism 4.0 software (GraphPad, San Diego, CA). The percentage effect data were calculated using Microsoft Excel, and visualizations were created using TIBCO Spotfire (TIBCO Software Inc., Palo Alto, CA). For the larger set of compounds, the individual percentage effect in the traditional capsaicin-based FLIPR assay and the various novel assays is plotted against each other to provide a qualitative assessment of the novelty of the hits that were identified. Correlations made between the novel FLIPR assays and the IonWorks-based voltage-ramp assay were calculated using the Spotfire simple linear regression tool. Here, the pIC50 values for the assay in question were plotted against the pIC50 values for the same compounds tested on the IonWorks platform, and the correlation coefficients were calculated by a least-squares method to give an indication of how closely the two data sets compared with one another.
Results
Gating Properties of TRPV1 Channels
The TRPV1 ligand-gated ion channel is a nonselective cation channel that can be activated by capsaicin, the pungent substance found in chili peppers, as well as low pH and heat >42 °C. Representative whole-cell patch-clamp recordings from the hTRPV1-CHO/Trex stable cell line used for all subsequent experiments are shown in
Voltage-Gated TRPV1 Pharmacology
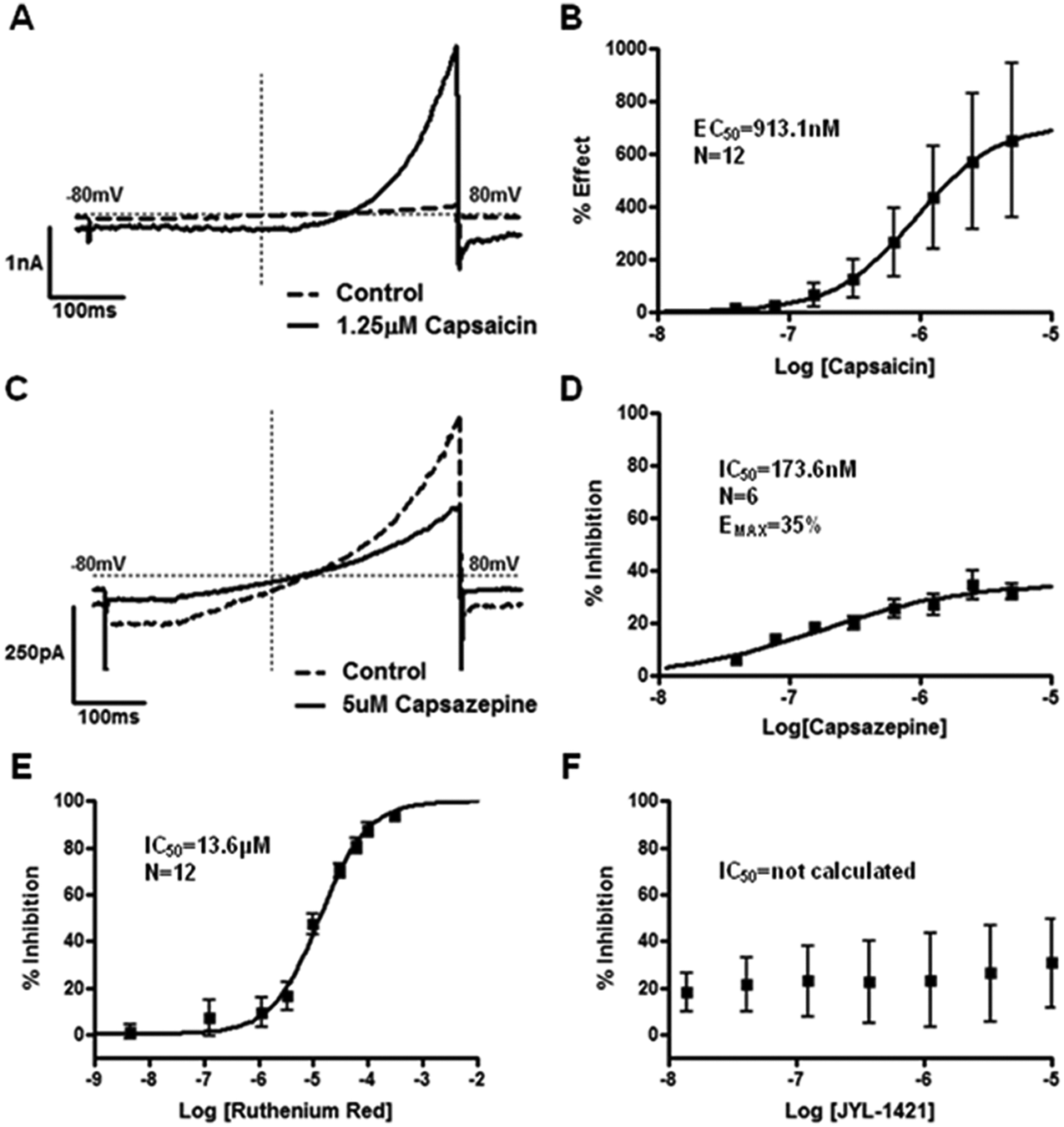
The pharmacology of commonly used TRPV1 antagonists and agonists was studied on the voltage-gated currents elicited by a voltage-ramp protocol that depolarized the cell from −80 mV to +80 mV in 500 ms using an IonWorks automated patch-clamp instrument. The protocol was identical to that used in the preceding manual patch-clamp experiments, except that the stimulation protocol was repeated every second for the duration of the experiment. After seals were made on the instrument and a perforated whole-cell access was achieved, each well on the IonWorks PatchPlate received a single concentration of a given compound, and responses were measured for 30 s. An example of the response to 1.25 µM capsaicin is shown in Figure 1a . The current measured at +80 mV was significantly greater in the presence of capsaicin as compared with the basal level, and the EC50 value for capsaicin was calculated to be ~1 µM ( Fig. 1b ).
(
Antagonists, in the absence of any exogenous ligand to stimulate TRPV1 channels, were also capable of blocking the outward current measured at +80 mV. Capsazepine, the prototypical TRPV1 antagonist, was able to block the outward current elicited by the voltage-ramp protocol ( Fig. 1c ). Although the effect was dose dependent, the highest concentration of capsazepine was not capable of completely blocking the voltage-gated TRPV1 current at +80 mV, with a maximal percentage inhibition of ~35% ( Fig. 1d ). Another reference TRPV1 antagonist, ruthenium red, was capable of completely blocking the voltage-gated currents with a calculated IC50 of ~13.6 µM ( Fig. 1e ). One of the few TRPV1 antagonists previously described to have differential pharmacology depending on the activation stimulus and species, JYL-1421,22–24 was also tested on the voltage-gated currents. Figure 1f shows that JYL-1421 was not capable of blocking the responses to the voltage-ramp protocol.
Development of Higher-Throughput Assays to Correlate to Voltage-Ramp Pharmacology
Automated patch-clamp instruments generate a wealth of data that are comparable with that of the gold-standard patch-clamp technique in a medium-throughput setting; however, they are still not capable of testing a very large set of compounds in a time- and cost-efficient manner. To circumvent this problem, three novel TRPV1 assays were developed and tested for (1) their potential to identify novel chemical entities compared with what more traditional capsaicin-based Ca2+ flux assays are capable of identifying and (2) their correlation with the voltage-gated pharmacology generated on the IonWorks assay. To develop these comparisons, two sets of compounds were tested on the novel assays. Set A contained 7509 compounds that represented ~75% of the diversity found within the AstraZeneca HTS library and adhered to Lipinski’s rules for compounds with druglike properties. 25 Set B contained 96 compounds that were known TRPV1 modulators, along with the compounds identified from each of the Set A screens with a percentage inhibition >50% from the novel assays.
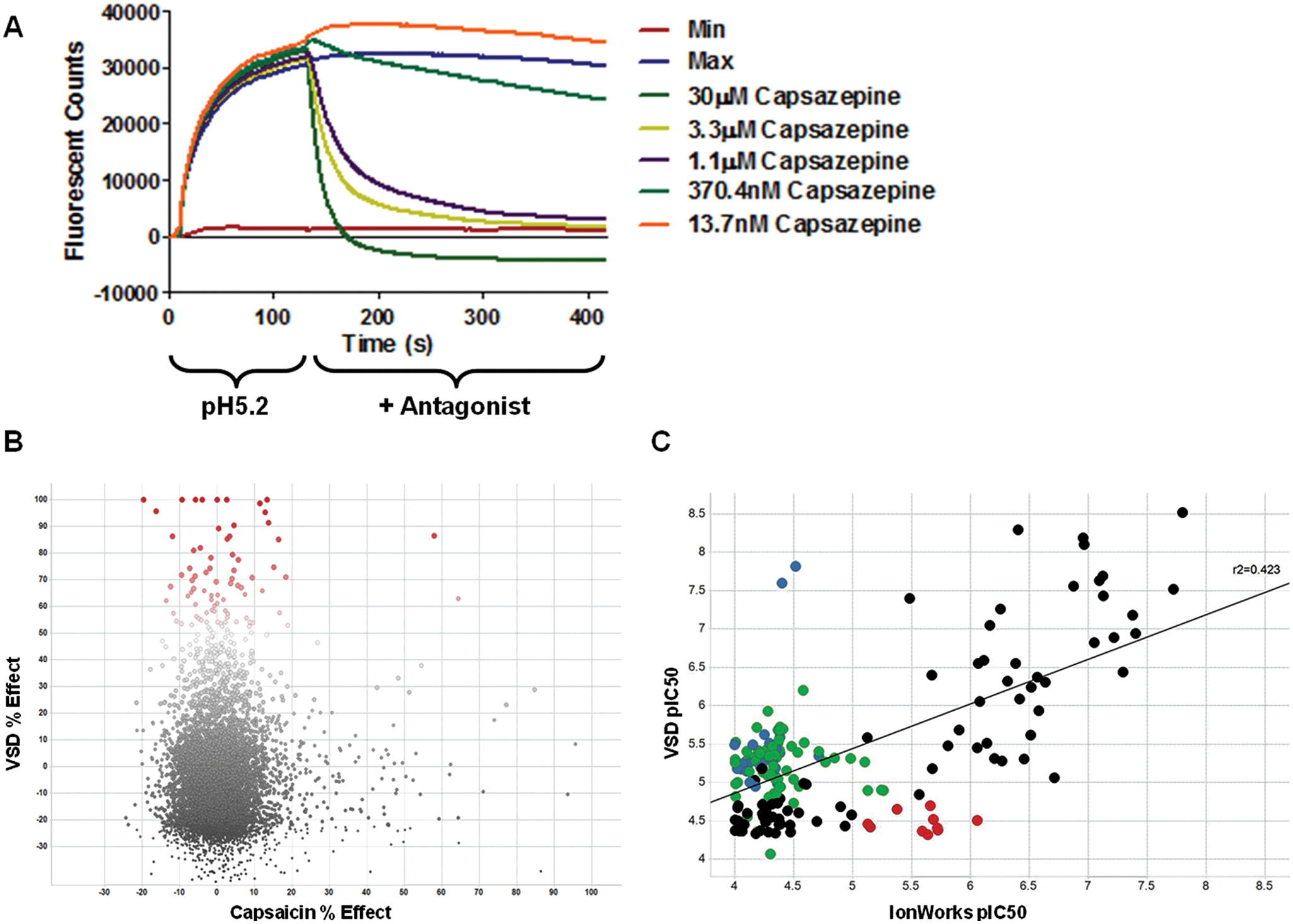
A voltage-sensitive dye-based assay was developed using pH 5.2 as a stimulus. The voltage-sensitive dye captures any changes to the resting membrane potential, and TRPV1 channels are activated prior to compound addition, representing a more physiological scenario when channels would be activated prior to compound being available. Figure 2a shows a typical example of the responses generated on the FLIPR 384-well instrument. The results from screening Set A with the voltage-sensitive dye assay are presented in Figure 2b . Of the 7509 compounds screened at a single concentration of 10 µM, 86 were found to have an inhibition >50%. Only two of these compounds were also found to have an inhibition >50% on the more traditional capsaicin-induced Ca2+-flux assay, indicating that the voltage-sensitive dye assay was capable of identifying TRPV1 antagonists with novel pharmacology. To assess whether the voltage-sensitive dye assay would be an appropriate HTS assay to reliably predict potency values on the voltage-gated IonWorks assay, the 86 novel compounds were included as part of the set B compounds that were tested in a full dose response in both assays. The correlation of pIC50 values obtained with the two methods is plotted in Figure 2c . The two assays had a significant correlation coefficient of r2 = 0.423; however, there were several compounds in the lower quadrants (red, VSD pIC50 < 5) that could potentially represent false-negatives. These compounds were not active in the voltage-sensitive dye assay but were considered active on the IonWorks assay. Conversely, there were also several compounds in the left-hand quadrants (blue, IonWorks pIC50 < 5) that were considered false-positives.
(
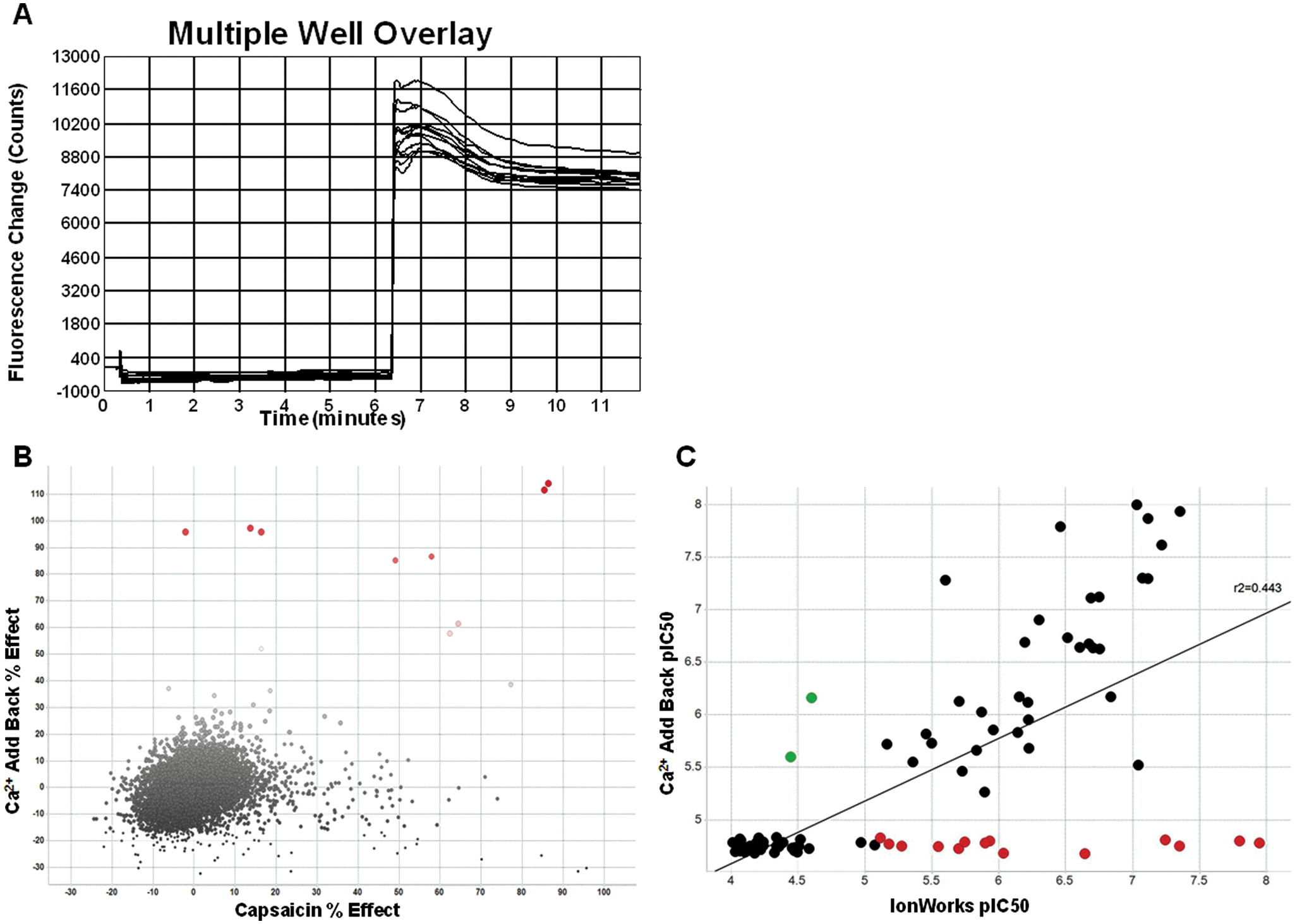
TRPV1 is sensitive to intracellular calcium concentrations, 26 and binding of Ca2+/calmodulin to the TRPV1 receptor plays an important role in channel desensitization after activation by capsaicin. Another TRP family member, TRPA1, has also been described to be sensitive to extracellular Ca2+ concentrations, where higher levels are capable of increasing channel gating. 27 We sought to determine if elevated extracellular Ca2+ concentrations could modulate TRPV1 in a similar fashion to TRPA1. Figure 3a shows the response of TRPV1-expressing cells to an elevated 4.8 mM Ca2+ concentration after being incubated in the presence of low 0.016 µM extracellular Ca2+. This response was not identified in nontransfected HEK293s cells nor in the noninduced hTRPV1-HEK/Trex cell line used for these experiments (data not shown). This calcium add-back assay was used in a similar screening paradigm as the voltage-sensitive dye method. Ten compounds were found to inhibit the Ca2+-induced responses >50% ( Fig. 3b ). Of these, five were previously identified using the traditional capsaicin-based FLIPR screen. Correlation of the data generated by the calcium add-back method to the voltage-gated IonWorks method using the set B compounds found a significant correlation coefficient of r2 = 0.443 ( Fig. 3c ). However, several compounds were found in the lower two quadrants of the correlation (red, CAB pIC50 < 5), including some with very high potency on the IonWorks assay (pIC50 > 7), indicating that the calcium add-back method might have the risk of generating too much false-negative data.
(
A third assay to screen for novel TRPV1 antagonists was developed using an atomic absorption spectroscopy-based rubidium flux method. Initially, the assay was developed to mimic the responses of TRPV1 channels to the voltage-ramp stimulus by adding a high concentration of KCl to depolarize the cells after loading with Rb+. Because cells normally treat Rb+ in a similar fashion to K+ ions, it was anticipated that measuring the effluxed Rb+ in the assay media after depolarization would give an indication of TRPV1 channel activation. Contrary to this expected response, no increase in Rb+ was detected in the extracellular assay media. While investigating the cause for this discrepancy, it was determined that TRPV1 antagonists such as ruthenium red and capsazepine were capable of inhibiting the uptake of Rb+ in TRPV1-expressing cells in a dose-dependent fashion. Although Rb+ is readily absorbed by nontransfected HEK cells in the absence of TRPV1, it appears as if Rb+ preferentially conducts through the TRPV1 channel when it is expressed, even in the absence of any other agonist to activate the channel. This unexpected response of TRPV1 channels offered an opportunity to assess novel TRPV1 pharmacology by measuring the amount of Rb+ capable of entering the cell after an antagonist preincubation. When screening the Set A collection of compounds, 39 compounds were found to inhibit Rb+ influx by >50% ( Fig. 4a ). Of these, only four were previously identified by traditional capsaicin-based screening methods. Comparing results from screening the set B compounds on both the atomic absorption method and the voltage-ramp method resulted in a correlation coefficient of r2 = 0.48 ( Fig. 4b ). Although several compounds were found in the lower-left quadrant that represent false-negative values (red, atomic absorption spectroscopy pIC50 < 5), these represented only ~27% of the total number of compounds, compared with ~55% in the voltage-sensitive dye method ( Fig. 2c ).
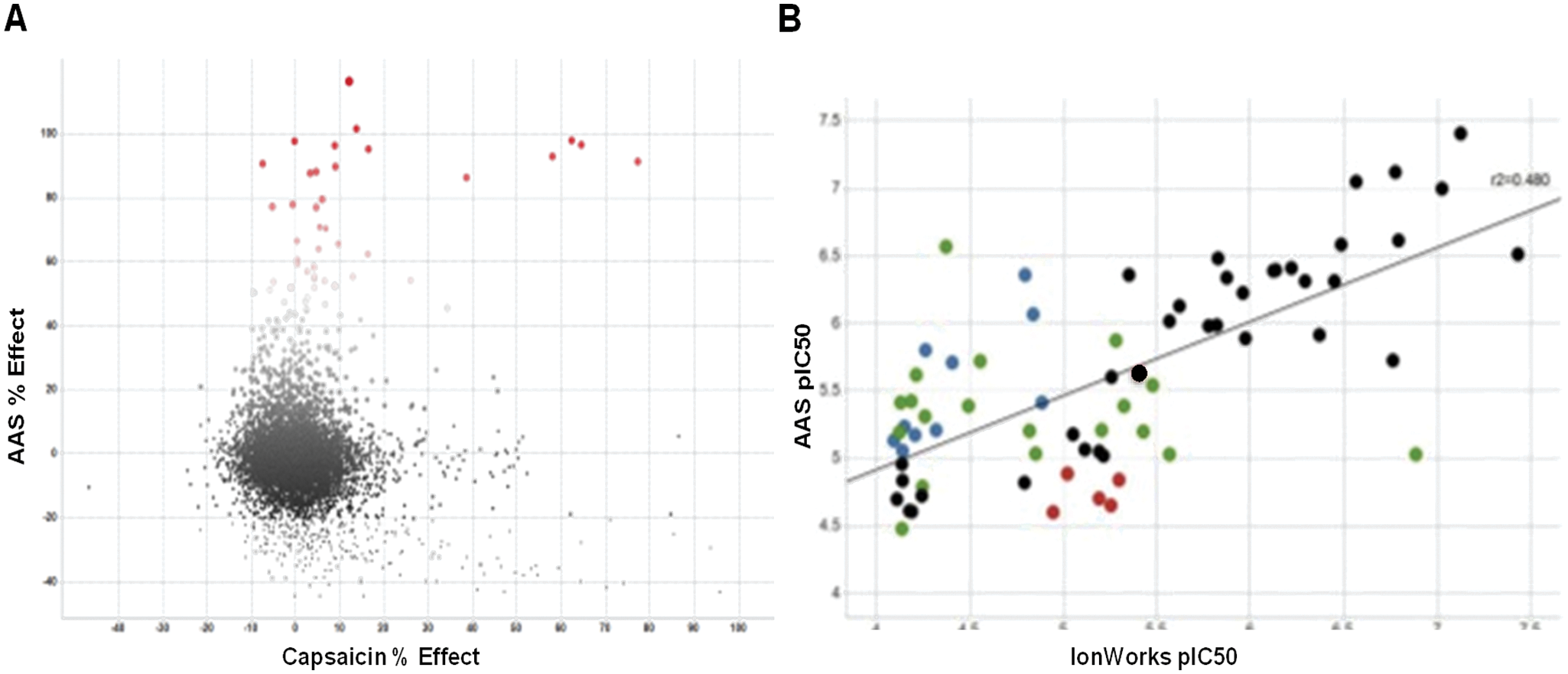
(
TRPV1 Antagonists Identified by Novel Screening Paradigm Independent of Capsaicin
Examples of the chemical properties for some compounds identified through the novel screening paradigms described above are listed in Table 1 . Several of these compounds were not previously identified with the more traditional capsaicin-induced Ca2+-flux assays and were some of the more potent molecules found with the novel assays. All compounds adhere to the Lipinski Rule of 5 in terms of druglike properties. 25 Two of the TRPV1 antagonists tested recently in clinical trials, AMG-51711 and SB-705498, 12 are included in the table for comparison. In general, solubility is improved for the novel compounds compared with these two clinical candidates. The pIC50 values from the traditional capsaicin-based FLIPR assay and the IonWorks voltage-gated assay can be found in the last two columns of Table 1 . It is interesting to note that several compounds exhibit a very different pharmacologic profile depending on the stimulus used to activate TRPV1 channels, and therefore the ability of a compound to inhibit the channel voltage-gated opening independent of any other agonist stimulation or shift in voltage dependence of activation should be added to the profiles of TRPV1 antagonists described previously.
Discussion
We have described the development of a novel assay to screen for TRPV1 antagonists using the previously reported voltage dependence for activation of the channel as a pharmacodynamic endpoint. Studying the pharmacology of known modulators on TRPV1 voltage-gated currents indicated that another dimension could be added to the previously described pharmacologic profiles for antagonists. 28 Although compounds have been differentiated before in terms of their ability to modulate capsaicin, low pH, and heat stimuli, 28 we have demonstrated that TRPV1 antagonists also possess differential effects on antagonizing agonist-independent voltage-gated responses of the channel. Some compounds such as ruthenium red are capable of fully antagonizing the voltage-evoked responses, whereas others such as capsazepine were very weak and partial antagonists.
Realizing this might offer new opportunities from a drug-discovery perspective, we sought to determine whether the voltage-gated activation of TRPV1 could be used to identify undiscovered chemical space in the otherwise relatively crowded patent literature 6 and improve on some of the chemical liabilities related to pharmacokinetic profiles and solubility that the majority of known TRPV1 antagonists possess. Although the pharmacodynamic effect on the voltage dependence of TRPV1 activation can be studied using automated patch-clamp instruments such as the IonWorks platform, this type of assay is not well suited for true high-throughput screening because its lower throughput and cost constraints. We therefore developed three novel assays capable of screening for TRPV1 antagonists independently of using capsaicin as an agonist to activate the channel. A medium-sized compound library was used to test each assay for its ability to identify new chemical starting points that had not been identified by more traditional capsaicin-based screening platforms.
Each of the three assays proved capable of finding such compounds, with the assays based on the voltage-sensitive dye detection of pH elicited responses and the spectroscopic atomic absorption of Rb+ retained in cells after spontaneous flux through TRPV1 giving the greatest number of new potential inhibitors (86 and 39, respectively). The ultimate goal was to return to the voltage-gated assays and compare these novel assays to determine which one would best predict the pharmacology on the automated patch-clamp assay. All three novel assays had similar correlation coefficients (r2 = 0.423 to 0.480). However, when looking at the distribution of the hits using TIBCO Spotfire visualization software, there was a very high incidence of false-negatives in the Ca2+ add-back assay. Although the goal of an HTS campaign is to trim the number of compounds down to a manageable set that can be profiled in greater depth, having the potential for too many false-negatives also runs the risk of not being able to identify the most ideal structures for future research. Furthermore, by screening the set of ~96 compounds, 11 of 26 compounds that had a pIC50 > 6 on the voltage-gated assay were not identified using the Ca2+ add-back HTS method, indicating that some highly potent antagonists could also be missed. In contrast, the voltage-sensitive dye method tended to identify several false- positive hits, with several compounds being inactive in the automated patch-clamp assay ( Fig. 2b ). The atomic absorption spectroscopy–based assays did not have the same issue of generating false-negative data compared with the Ca2+ add-back assay, had fewer than the voltage-sensitive dye assay, and had the best correlation with the automated patch-clamp assay. The atomic absorption spectroscopy–based assay was able to positively identify 11 of 12 compounds that also had a pIC50 > 6 on the voltage-gated assay compared with 17 of 26 with the voltage-sensitive dye method.
Several compounds were identified by these novel assays that had previously not been identified in capsaicin-based HTSs. Therefore, after their identification, they were further characterized using the more traditional capsaicin-based FLIPR assay to verify their potency. Data in Table 1 indicate that although some of the compounds were first identified by the novel assays described here, they still maintained a good potency in the capsaicin-based assay. However, several compounds were characterized by a significant difference in terms of the potency to inhibit the voltage-gated activation of TRPV1 compared with the capsaicin activation. These represent a novel profile of TRPV1 antagonists that should be added to those described previously. 28 The differences and relationship of the novel pharmacology identified using the three novel screening assays can be visualized in the Venn diagram presented in Figure 5 . Each assay was capable of identifying supposed inhibitors of TRPV1 using their respective endpoint measures; however, there is very little overlap between these novel pharmacologies.
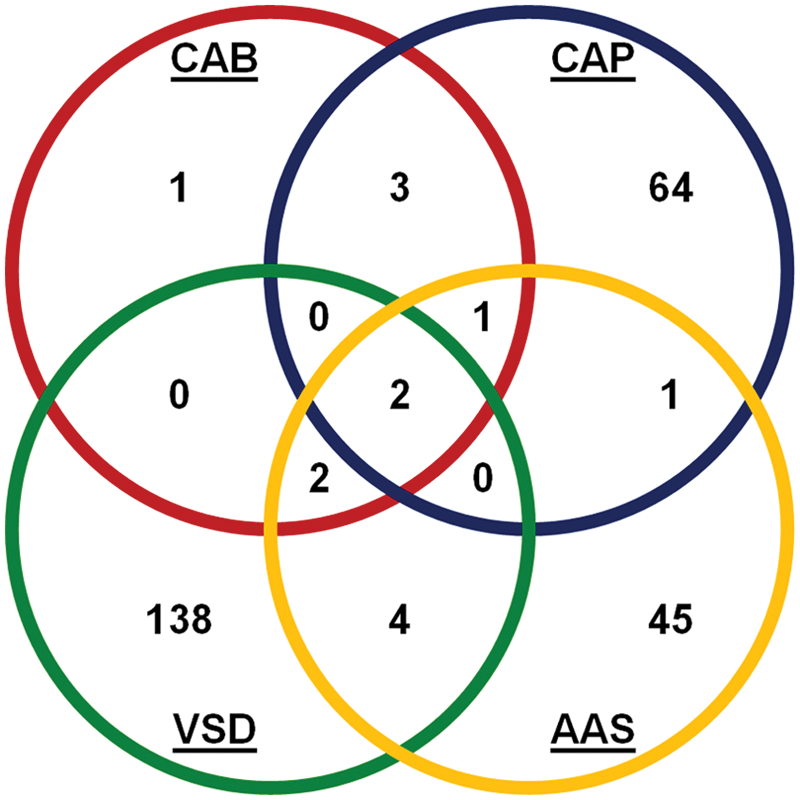
Venn diagram to highlight the differences and similarities in the compounds identified by the three novel assays and the traditional capsaicin-based assay. CAPS = capsaicin-based assay; VSD = voltage-sensitive dye–based assay; CAB = Ca2+ add-back assay; atomic absorption spectroscopy = the atomic absorption spectroscopy–based Rb+ flux assay.
The voltage-gated activation of TRPV1 is not a novel concept. In fact, it has been well documented that while capsaicin and resiniferatoxin reveal a voltage-independent gating for TRPV1, the more natural ligands such as protons (low pH) and heat shift the voltage dependence of gating to more physiological negative potentials.13,29 An allosteric model to describe the synergy between voltage gating and other independent stimuli to regulate channel gating has already been described. 30 Several molecular determinants responsible for the proton- and heat-dependent shifts in the voltage dependence of activation have been described, including F660 31 in the S6 transmembrane domain and I696, W697, and Q700 in the TRP box. 32 It would be interesting to assess the types of molecular interactions the compounds identified here have with TRPV1 and determine if they have a better profile than some of the previous TRPV1 antagonists, particularly with respect to causing hyperthermia in vivo. 33 In addition, because TRPV1 is hypothesized to be a target for the treatment of chronic inflammatory pain, the analgesic potential of these compounds should be investigated and will be the subject of future research.
Footnotes
Acknowledgements
The authors would like to thank Jennifer M. A. Laird for reviewing the content of the article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
All authors were employed by a for profit company, AstraZeneca, at the time when experiments were conducted.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.