
Abstract
The aim of this study was to demonstrate proof-of-concept feasibility for the use of human neural stem cells (NSCs) for high-throughput screening (HTS) applications. For this study, an adherent human induced pluripotent stem (iPS) cell–derived long-term, self-renewing, neuroepithelial-like stem (lt-NES) cell line was selected as a representative NSC. Here, we describe the automated large-scale serum-free culture (“scale-up”) of human lt-NES cells on the CompacT SelecT cell culture robotic platform, followed by their subsequent automated “scale-out” into a microwell plate format. We also report a medium-throughput screen of 1000 compounds to identify modulators of neural stem cell proliferation and/or survival. The screen was performed on two independent occasions using a cell viability assay with end-point reading resulting in the identification of 24 potential hit compounds, 5 of which were found to increase the proliferation and/or survival of human lt-NES on both occasions. Follow-up studies confirmed a dose-dependent effect of one of the hit compounds, which was a Cdk-2 modulator. This approach could be further developed as part of a strategy to screen compounds to either improve the procedures for the in vitro expansion of neural stem cells or to potentially modulate endogenous neural stem cell behavior in the diseased nervous system.
Introduction
Multipotent self-renewing mammalian, including human, neural stem cells (NSCs) have been enriched and expanded in vitro from central nervous system (CNS) tissue and pluripotent embryonic stem cell sources, either as self-adherent aggregates forming neurospheres in suspension or as adherent monolayers on substrate-coated tissue culture plates.1,2 Human NSCs and their derivatives hold great potential for regenerative medicine, both as direct therapeutics, as well as an enabling technology for the discovery and development of conventional small-molecule drugs for the treatment of CNS disorders. 3
A key area of CNS therapeutic interest is neurogenesis, and although the generation of new neurons in the mature CNS was first demonstrated in 1965, it was only 40 years later that adult neurogenesis became an accepted phenomenon. 4 The process of neurogenesis can be considered as a sequence of component events that includes the proliferation of neural stem and/or progenitor cells in the CNS and their subsequent migration, differentiation, maturation, and integration. Although the biological significance of adult neurogenesis remains unclear, it is likely that some or all of the component events may be beneficial to achieve some degree of restoration and functional recovery following neurodegenerative insult. Two restricted neurogenic niches persist into adulthood—namely, the subventricular zone (SVZ) of the lateral ventricle and the dentate gyrus subgranular zone of the hippocampus (reviewed by Alvarez-Buylla and Lim 5 ). Interestingly, established antidepressant drugs such as the serotonin reuptake inhibitor fluoxetine (Prozac) were recently confirmed to exert stimulatory effects on neurogenesis in the adult dentate gyrus 6 and to increase the viability and proliferation of hippocampus-derived NSCs in vitro. 7
Accordingly, a future therapeutic goal may be to develop small-molecule modulators of resident adult NSC to facilitate or to enhance repair following acute traumas, such as head injuries and strokes, or as a treatment for neurodegenerative diseases and depression. A practical starting point for the development of such therapeutics is the screening of small molecules for their potential to modulate the expansion and/or differentiation of adult NSCs in vitro. Ultimately, there is also the therapeutic potential for such small molecules to be used in conjunction with an NSC transplantation modality.
To perform a drug compound screen, an NSC line with suitable biological properties that can be sufficiently expanded in vitro for the high-throughput screening (HTS) campaign is required. Ideally, the NSC line is amenable to automation of cell culture scale-up and scale-out into screening ready microwell plates. In fact, a robust and scalable cell line and in vitro cell culture process are the first of three key requirements for a successful industrial cell-based HTS campaign. In this regard, a paradigm shift of the stem cell field occurred with the discovery of somatic cell reprogramming, whereby expression of the transcription factors Oct4, Sox2, c-Myc, and Klf4 was shown to induce fibroblasts to become pluripotent stem cells (designated as induced pluripotent stem [iPS] cells). 8 This discovery has two major applications for the field of regenerative medicine: first, the potential to supply large numbers of autologous cells for transplantation that are capable of differentiation into all cell lineages and, second, the derivation of disease-specific stem cells and panels of stem cells of diverse genetic backgrounds for drug discovery and safety screening (reviewed by Csete). 9 These unique properties provided the motivation to explore and confirm the applicability of protocols that were devised to derive long-term stably proliferative neuroepithelial-like stem (lt-NES) cells from pluripotent human embryonic stem cells 10 to human iPS cells.11,12 The human lt-NES cell line used in this study was derived in two stages; human fibroblasts were first reprogrammed into iPS cells, and then the human iPS cells were subsequently differentiated into stably proliferative lt-NES. 11
Human lt-NES cells of embryonic stem (ES) and iPS cell origin are routinely expanded as an adherent monolayer culture in serum-free media conditions supplemented with epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF2). Upon growth factor withdrawal, these human lt-NES cells differentiate to give rise to a mixed population of cells containing neurons, astrocytes, and oligodendrocytes.10,11 As proof of principle for the concept of disease modeling with lt-NES of iPS cell origin, a disease-specific protein aggregation phenotype in neurons differentiated from lt-NES, which were derived from iPS cell lines generated from the fibroblasts of patients with Machado-Joseph disease, was demonstrated. 12 Even though a recent transplantation study of human iPS cell-derived lt-NES in a mouse model of spinal cord injury reported functional recovery of hind limb motor function, 13 this is just a first glimpse of hope for potential cell therapy applications. In comparison, there is already a large body of evidence, accumulated over a decade, for the efficacy of human CNS-derived NSCs in preclinical rodent models of disease or injury in all of the three structures comprising the CNS—the brain, the spinal cord, and the retina14–16—along with their successful translation into clinical trials (www.clinicaltrials.gov; trial identifiers: NCT00337636, NCT01005004, NCT01321333). It therefore follows that disease modeling and drug screening represent more immediate opportunities for human iPS cell-derived lt-NES cells to be used in the discovery of new medicines and, hence, our interest in their application in this study.
The second requirement for a successful cell-based screening campaign is a robust cell-based assay that is amenable to miniaturization such that the assay can be exploited in a high-throughput manner. Primary mouse NSCs derived from the SVZ and grown as neurospheres have previously been used by Liu et al. 17 for the development of small-well format cell-based assays for HTS of a 1.4 million small- molecule pharmaceutical compound collection. In the latter case, a homogeneous adenosine triphosphate (ATP)–bioluminescence–based cell viability assay, which equates the amount of intracellular ATP to viable cell numbers, was employed since it is extremely sensitive and can detect changes to total cell number at a density of 50 cells per well in a 96-well plate. 17 Miniaturization of cell-based assays, including the above-described ATP-bioluminescence–based assay, has also been demonstrated using stably proliferative adherent mouse NSCs in a range of microwell plate formats. 18 Based on previously demonstrated adequate performance,17,18 we initially chose the ATP-bioluminescence–based cell viability assay to perform a proof-of-concept screen.
The third requirement for a successful cell-based assay validation for HTS is to have an appropriate library of compounds to test. For our study, a set of 1000 compounds was prepared by Summit plc (Oxford, UK). The library comprised compounds with known pharmacology/mode of action along with “new chemical entities” (NCEs) and had previously been screened using adherent mouse NSCs and the ATP-bioluminescence–based cell viability assay format. 19
The objectives of this study were twofold: first, to develop protocols for the automated maintenance and scale-out of human lt-NES cells and, second, to develop and validate an assay for screening 1000 small-molecule compounds for effects on human lt-NES cell survival and/or proliferation. Herein we report how protocols for the automated long-term maintenance of human lt-NES cells on the CompacT SelecT automated cell culture platform and a manually plated ATP-bioluminescence–based cell viability assay were developed and applied to achieve these two objectives.
Materials and Methods
Routine Human lt-NES Cell Culture
Human AF22 iPS cell-derived lt-NES cells11,20 were cultured according to previously described methods,10–12 with minor modifications. Cells were maintained in RHB-Basal media (#SCS-SF-NB-00; StemCells, Inc., Newark, CA) supplemented with NDiff N2 (#SF-NS-01-005; StemCells, Inc.) at a concentration of 1:200 and B27 (#17504-044; Invitrogen, Carlsbad, CA) at a concentration of 1:1000, 10 ng/mL EGF (#100-15; Peprotech EC Ltd., London, UK), and 10 ng/mL FGF2 (#100-18-B; Peprotech EC Ltd.). Flasks and plates were coated with 0.01% polyornithine (#P4957; Sigma, St. Louis, MO) for 30 min at 37 °C followed by two phosphate-buffered saline (PBS) washes and treatment with 10 µg/mL laminin (#23017-015 diluted in PBS; Invitrogen) for a minimum of 3 h at 37 °C. Cells were passaged every 2 to 3 days using 0.05% trypsin (#25300; Invitrogen) to release the cells from the flasks followed by treatment with trypsin inhibitor (#R-007-100; Invitrogen) prior to centrifugation and resuspension in fresh media. Cells were seeded at a density of 4 × 104 cells/cm2 in T25 or T75 flasks.
The CompacT SelecT Automated Cell Culture Platform
The CompacT SelecT (TAP Biosystems, Royston, UK) is capable of the expansion and maintenance of multiple cell lines, cell counting and viability measurements, and the harvesting and plating of cells for assays and screening. It has the capacity for the incubation of up to 130 T175 flasks and 210 microwell plates and houses a sterile class II biosafety cabinet to ensure that all manipulations are performed in an aseptic manner. A hydraulic robotic arm manipulates barcoded flasks and liquid pipetting heads to perform the cell culture manipulations, and 10 in-line pumps are available for media/reagents that can be stored at 2 to 8 °C or at room temperature as required.
Automated Scale-up and Scale-out of Human lt-NES Cells
Cells were seeded at a density of 8 × 106 per T175 flask (BD #353118, Franklin Lakes, NJ). T175 flasks were precoated with polyornithine and laminin (as described for routine culture). RHB-Basal media (#SCS-SF-NB-00; StemCells, Inc.) was combined with NDiff N2 (#SF-NS-01-005; StemCells, Inc.) and plumbed onto the CompacT SelecT and stored at 2 to 8 °C. Then, 0.05% trypsin (#25300; Invitrogen) and trypsin inhibitor (#R-007-100; Invitrogen) were both plumbed in and stored at 2 to 8 °C. Additions of B27, EGF, and FGF2 were made using the CompacT SelecT’s automated pipetting head on a daily basis via a static tube that was manually inserted into the biosafety chamber. The cells were passaged every 2 to 3 days following visual inspection of confluency. The detail of the automated protocol is described in the Results section.
Cell Viability Assay and Medium-Throughput Screening
We quantified relative human lt-NES cell proliferation and/or survival using a cell viability assay that has been shown previously to correlate directly with both the proliferative activity of, and the BrdU incorporation into, neural stem/progenitor cells. 21 The activity of hits that are identified and predicted to affect neural stem cell proliferation and/or survival can then be confirmed using classical methods. The ATP-bioluminescence cell viability assay (#G7570; Cell Titer-Glo, Promega, Madison, WI) is a fast and convenient method for monitoring relative cell viability in a microwell plate format, making it a suitable method for primary screening because the homogeneous assay procedure involves the addition of a single reagent directly to the cells. 17 White flat-bottom Nunc 96-well plates (#136101; ThermoFisher, Waltham, MA) were pretreated as for manual culture with 0.01% polyornithine (#P4957; Sigma) 50 µL/well for 30 min at 37 °C, followed by 2× PBS washes, and 10 µg/mL laminin (#23017-015; Invitrogen) 50 µL/well for a minimum of 3 h (plates were routinely treated with polyornithine and laminin the day before the assay was set up; laminin was aspirated immediately prior to assay setup). On day t = 0, assay plates were set up with growth factor (GF) controls/test compounds prior to addition of 2000 cells/well to a final volume of 100 µL/well. Cells in the control wells were plated either in the absence of EGF and FGF2 (no GF = negative control) or in the presence of maximal EGF (10 ng/mL) and FGF2 (10 ng/mL) (max GF = positive control). All controls were tested in the presence of 0.1% DMSO, which is equal to the background concentration in which the compounds were tested. Assay plates were placed within gas-permeable humidity chambers (Perspex boxes containing sterile dH2O-soaked absorbent towels) to eliminate plate edge effects and incubated for 96 h in 5% CO2 in air at 37 °C. Providing that assay linearity in relation to cell density is first validated, the Cell Titer-Glo reagent can be diluted beyond the manufacturer’s recommendations, thereby reducing screening costs and also reducing potential cross-talk effects. For this study, the prepared Cell Titer-Glo reagent was diluted a further 1:11 in PBS + 0.2% Triton X-100; following this dilution, the assay was performed as per the manufacturer’s instructions and total luminescence read on the EnVision Multilabel Plate Reader (PerkinElmer, Waltham, MA). Compound screening was performed in the absence of EGF and FGF2, with all compounds being tested at a final concentration of 1 µM in a background concentration of 0.1% DMSO. Screening data are normalized to the negative control (no GFs) on a plate-by-plate basis and then represented as fold change relative to the negative control. To test the robustness and reproducibility of the screen, Z′ factor values were calculated from the positive and negative control values for each assay plate according to the methods of Zhang et al. 22 For dose-response follow-up studies, data are normalized to the positive control (max EGF + FGF2) on a plate-by-plate basis and then represented as % activity relative to the positive control. For both the initial screens and for the dose-response follow-up studies, compounds were routinely diluted in cell culture media to an intermediate concentration and then manually added to the assay plates in a volume of 30 µL followed by addition of cells in a volume of 70 µL. All manipulations were performed manually using handheld semiautomated multichannel pipettes.
Compound Library
A selection of 1000 compounds from Summit plc’s 50 000-member screening collection was prepared for this screening campaign containing two subgroups. One subgroup comprising 600 compounds was selected from Summit’s 3000-tool compound collection; these are compounds with known pharmacology, off-patent drugs, clinical failures, natural products, and so on. Since these compounds have known pharmacology, they are useful to help elucidate potential targets and modes of action that cause changes in neural stem cell fate. The second subgroup comprised 400 NCE compounds selected based on their previous identification as primary hits when Summit performed a 10 000-compound NCE screen against Drosophila neuroblasts to identify compounds affecting asymmetric division. The compound library was supplied in a 96-well format at 10 mM in DMSO. Dose-dependent follow-up studies were performed using freshly dissolved lots of the 24 hit compounds, supplied in glass vials at 10 mM in DMSO.
Results
Automated Scale-up and Scale-out of Human lt-NES Cells
Human lt-NES cells in adherent culture are routinely passaged using trypsin followed by treatment with a trypsin inhibitor and removal of both agents by centrifugation. Hence, the development of automated culture protocols for human lt-NES cells on the CompacT SelecT robotic cell culture platform required modifications to the routine passage methods because the CompacT SelecT does not have an integral centrifuge. Since trypsin is a relatively harsh dissociation reagent, initial studies focused on the evaluation of alternative cell dissociation reagents (e.g., Accutase and Cell Dissociation Buffer), since these have been successfully employed in previous automation campaigns for both murine and human brain tissue–derived, adherent NSCs.19,23 However, under the conditions evaluated, neither Accutase (#L11-007; PAA Laboratories GmbH, Pasching, Austria) nor Cell Dissociation Buffer (#13151-014; Invitrogen) proved to be effective at passaging the human lt-NES cells; we think this may be due to the fact that human lt-NES cells are likely very firmly adhered as they are routinely being expanded on a substrate of polyornithine combined with laminin.
Attempts were then made to develop a protocol in which the trypsin and trypsin inhibitor would simply be diluted as a natural consequence of the passage procedure. However, the results showed that the human lt-NES cells would not tolerate residual trypsin and trypsin inhibitor; therefore, this strategy was abandoned. Ultimately, an approach was developed in which the trypsin and trypsin inhibitor were removed following the passage step by performing a full media change after the majority of the cells had been allowed to attach sufficiently to withstand the media change. The automated protocol was further modified by increasing the plating density to account for the subsequent media change. We also investigated whether there was any advantage to prewarming the media and/or enzymes immediately prior to the passage process. For many cells, prewarming reagents does not confer a worthwhile advantage over the yield generated using cold reagents; however, for the human lt-NES cells, we found that the cells had an improved yield in an automated setting if the reagents were prewarmed for use in the passage procedure (data not shown). With these final refinements, the human lt-NES cells displayed comparable morphology following expansion on the CompacT SelecT automated cell culture platform ( Fig. 1B ) when compared with manually cultured controls ( Fig. 1A ). Furthermore, when compared, the cumulative doubling rate of human lt-NES cells maintained on the CompacT SelecT exceeded that of a culture that had been manually maintained ( Fig. 2 ). Because the software on the CompacT SelecT is designed for more routine tasks, it was necessary to program the automation protocol in a stepwise fashion. Hence, the complete automated culture process for human lt-NES cell passage on the CompacT SelecT platform comprises six separately programmed steps that are briefly outlined as follows: (1) Dump laminin from pretreated flasks; (2) prewarm trypsin and apply to cells; (3) prewarm media and harvest cells; (4) incorporate laminin-coated flask as “last used”; (5) count cells, seed at 8 × 106 into a laminin-coated flask, pipette B27 + EGF + FGF2 from static tube into culture flask, and incubate flask; and (6) 2 h postpassage (i.e., after cells have moderately adhered to flask), perform complete media change and supplementation with B27, EGF, and FGF2 from the static tube.
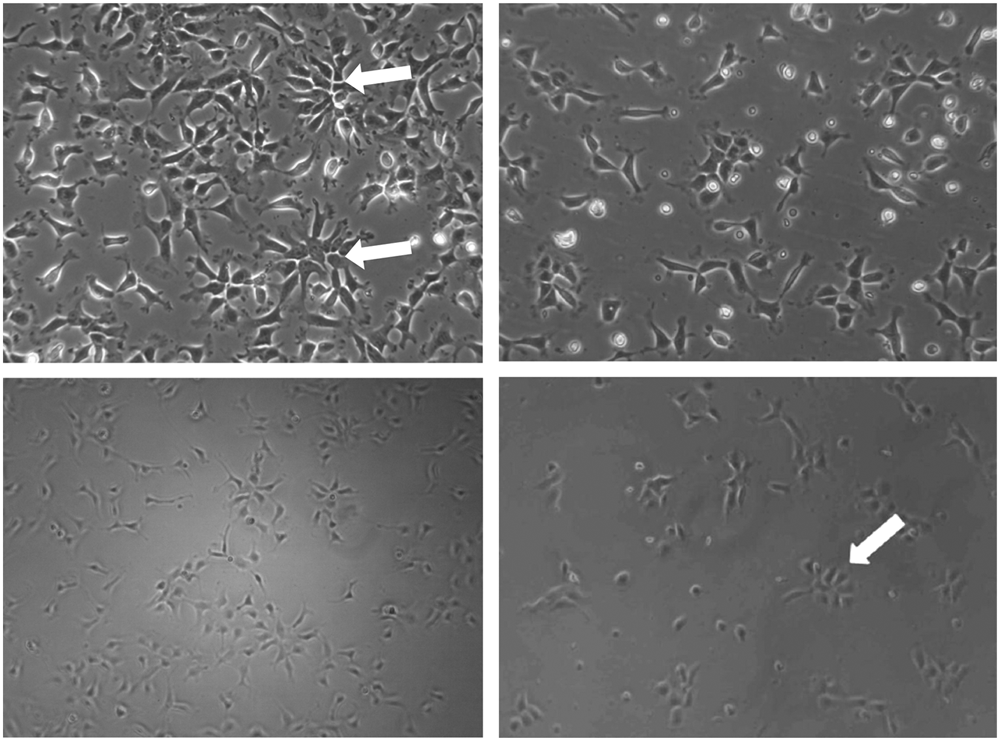
Development of automated protocols for scale-up and scale-out. The morphology of manually cultured human neuroepithelial-like stem (lt-NES) cells in T25 flasks is shown in
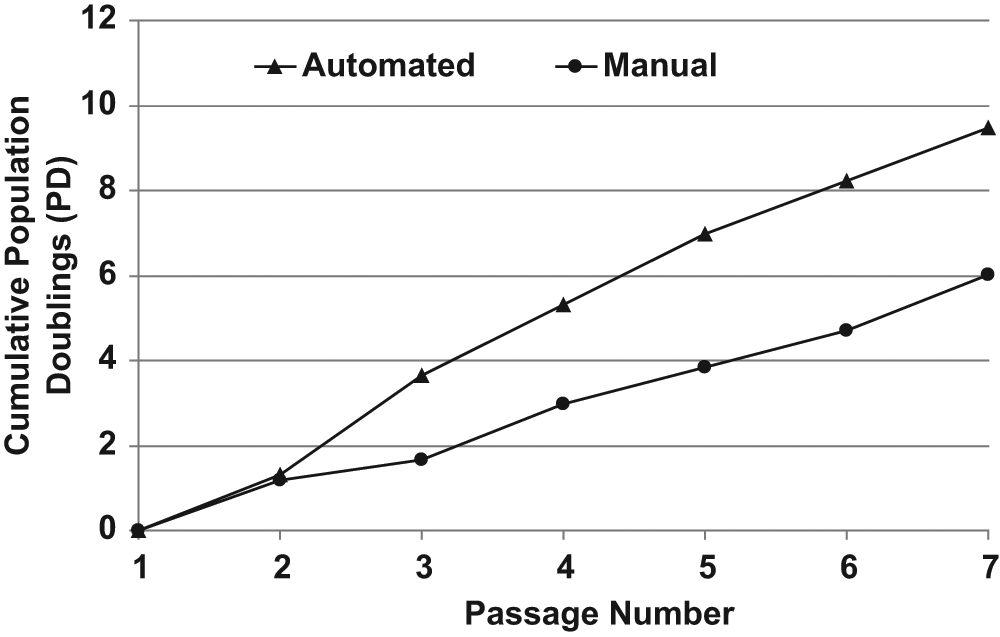
Development of automated passage protocol. Comparison of cumulative population doublings of the human induced pluripotent stem cell–derived neuroepithelial-like stem (lt-NES) AF22 cell line that was either maintained on the CompacT SelecT (“Automated,” triangles) or manually (“Manual,” circles). The passage numbers indicated refer to the number of actual automated or manual passages, which corresponds to an overall passage range of P57–63 for the “Automated” data and P51–57 for the “Manual” data.
Scale-out plating of the human lt-NES cells into a 96-well plate format employs the same first three steps used for maintenance of the cells above. At step 4, the process diverges and the cells are prepared at a density suitable for seeding into 96-well plates. Two to 3 h after plating cells into 96-well plates, a manual media change has been employed to remove residual trypsin because the plating module of the CompacT SelecT used for this study does not have the capacity to aspirate media from 96-well plates. Therefore, we have performed the media change manually to demonstrate the potential for a fully automated maintenance and scale-out protocol. However, in order for the scale-out to be fully automated, a cell feeder would need to be employed to perform the media change 2 to 3 h postplating. There is the potential to build an automated cell-feeding liquid-handling module into the CompacT SelecT, which would greatly enhance applications for cells differentiating in 96- and 384-well formats. In the absence of such an integrated system, the use of an offline liquid-handling system would still enable the procedure to be performed in an automated fashion. Using the methods described above, human lt-NES cells have been successfully plated into 96-well plates at 6 × 103 cells per well using the CompacT SelecT automated cell culture platform, and as shown in Figure 1D , the morphology of the cells after an offline media change and incubation in maintenance media overnight is indistinguishable from that of manually plated cells in 96-well plates (shown in Fig. 1C ). Furthermore, the cells stain homogeneously positive for the NSC markers Sox2 and Nestin (data not shown). When working to capacity, with the protocols that we have developed, the CompacT SelecT is capable of generating over 400 × 96-well or 384-well plates of lt-NES cells on a weekly basis, a capacity sufficient to furnish an HTS campaign.
Development of Human lt-NES Cell Viability Assay
The human lt-NES cell viability assay was optimized by testing a range of assay conditions, including cell density and time course to achieve the greatest signal to noise between positive and negative controls (data not shown). Cells plated in the absence of growth factors (EGF + FGF2) served as negative controls, and positive controls were cells plated in the presence of EGF + FGF2 at the concentrations normally used for maintenance (10 ng/mL EGF + 10 ng/mL FGF2). All manipulations (plating of cells, media, compounds and assay reagents) were performed manually using handheld semi-automated multichannel pipettes. To validate the assay and confirm that it is capable of detecting an increase in proliferative rate in response to varying concentrations of growth factors, total cell viability in response to EGF or FGF2 was evaluated, and a dose-dependent effect was demonstrated in response to FGF2 ( Fig. 3 ). Prior to the performance of the screen, possible edge effects were assessed by measuring uniformity of signal over the whole plate for 0% (no cells) and 100% (2000 cells/well) with (GF) or without growth factor (no GF) conditions over the 4-day assay duration. The four corner wells were found to be most vulnerable to edge/evaporation effects. This issue was addressed by the incorporation of humidity chambers, which reduced the standard deviation (SD) over the whole plate from 0.191, 0.197, and 0.207 to 0.146, 0.146, and 0.066 for the 0%, 100% (no GF), and 100% (GF) conditions, respectively. In addition to reducing the overall SD, utilization of humidity chambers almost completely resolved the issue of the previously observed microplate edge evaporation assay distortions.
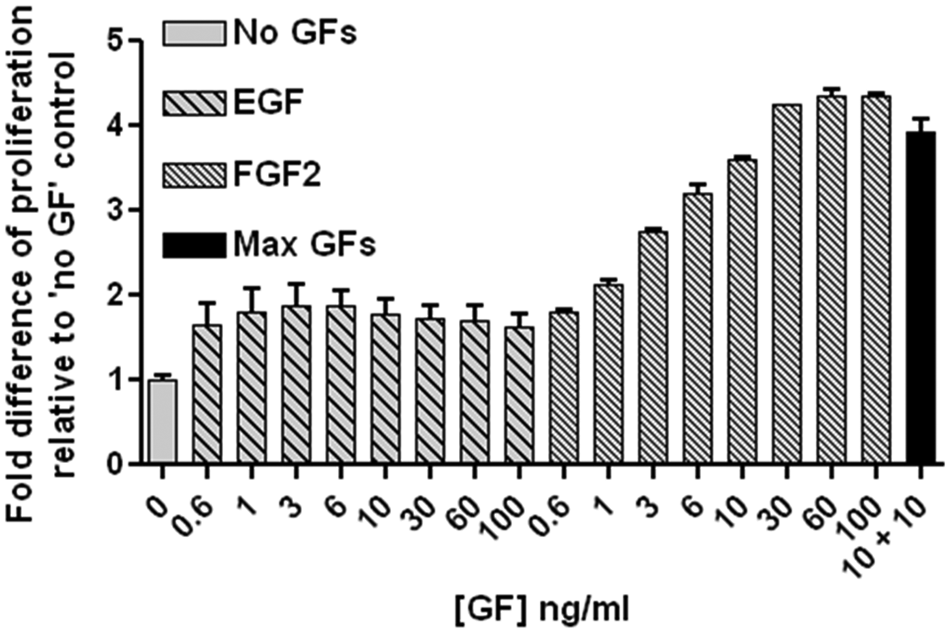
Adenosine triphosphate (ATP) cell viability proliferation/survival assay. Growth factor (GF) dose responses were evaluated by plating 2000 neuroepithelial-like stem (lt-NES) cells per well in a 96-well plate format and performing the ATP-bioluminescence cell viability assay after 4 days of incubation. Data are presented as “fold difference” of luminescent signal relative to negative control. The assay is capable of detecting a dose-dependent increase in cell proliferation with increasing concentrations of fibroblast growth factor 2 (FGF2; series of hashed bars to the right) relative to the negative control of “no growth factors” (solid gray bar). In contrast, increasing concentrations of epidermal growth factor (EGF) do not elicit a dose-dependent increase in proliferation (series of hashed bars to the left). The positive control (solid black bar) represents the condition in which the cells are routinely maintained (10 ng/mL EGF + 10 ng/mL FGF2) and the signal to noise of approximately 4 to 1 represents the expected doubling rate of the lt-NES cells in 4 days.
Human lt-NES Cell Medium-Throughput Screening of 1000-Compound Library and Dose-Response Follow-up Studies
The human lt-NES cell proliferation and/or survival screen of the 1000 compounds was performed in a 96-well plate format with duplicate wells positioned on sister plates, and the complete screen was performed on two independent occasions. There was an acceptable signal-to-noise window between the positive and negative controls on a plate-by-plate basis, with a mean fold difference of 4.24 ± 0.10 (mean ± SEM) for screen 1 and 4.52 ± 0.10 for screen 2 ( Fig. 4A ). The signal-to-noise window was deemed acceptable because a fourfold increase in total cell number over 4 days was in line with the expected proliferation rate of the lt-NES cells when grown under standard conditions in the presence of EGF + FGF2. Furthermore, Z′ factor values were observed to be relatively high for a cell-based assay with a 4-day incubation period. Z′ factor values were calculated to be 0.67 ± 0.02 (mean ± SEM) for screen 1 and 0.72 ± 0.02 for screen 2 (n = 26 plates for each screen), demonstrating the robustness of the assay. 22 Hits were defined as compounds that stimulated an increase in cell survival/proliferation in the absence of growth factors above a threshold value that was specific for each of the two screens (the second method being introduced as an improvement). Threshold values were calculated as deviations of two or three SDs above the mean values of the negative control data points for the whole screen (screen 2) or deviations of two or three SDs above the mean values of the 1000-compound data set with toxic compounds excluded (screen 1, with toxic compounds being defined arbitrarily as compounds that reduced cell viability to less than 50% of the negative control).
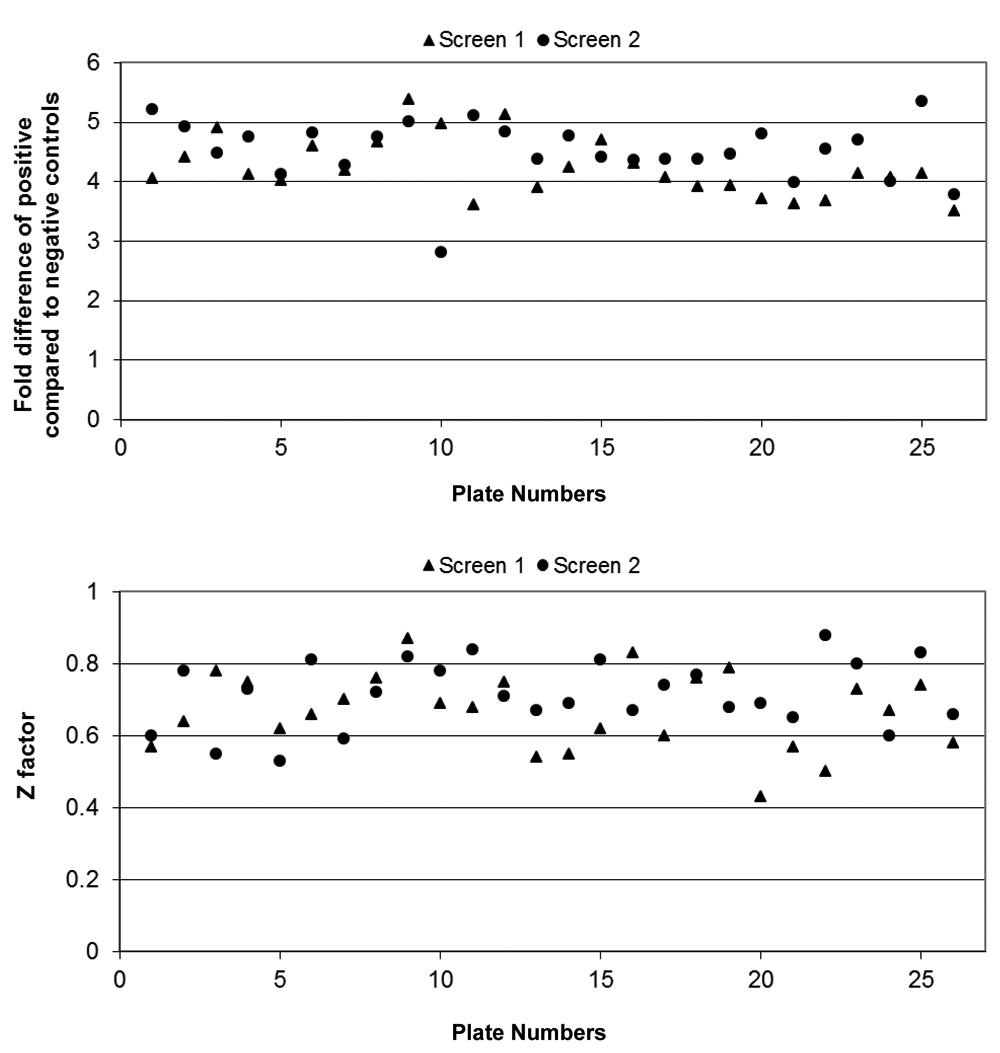
Screen performance. Signal to noise was monitored by comparing the mean positive control value to the mean negative control value on a plate-by-plate basis for each plate of the two independent screens and plotted as “fold difference.” The mean fold difference was 4.24 ± 0.10 (mean ± SEM) for screen 1 and 4.52 + 0.10 (mean ± SEM) for screen 2. Individual fold difference values are plotted in
The 1000-compound library (provided by Summit plc.) included 600 “tool compounds” with known modes of action in addition to 400 NCEs previously shown to affect asymmetric division of Drosophila neuroblasts, thereby representing potential start points for a medicinal chemistry optimization program. The 1000-compound screen was performed in the absence of EGF and FGF2, with all compounds being tested at a concentration of 1 µM, and a total of 24 compounds were identified that showed increased cell proliferation/survival. Of those 24 primary hits, 21 were from the tool compounds collection and 3 from the NCE collection. Seven of the 24 compounds were also identified in a previous screen of mouse neural stem cells with the same 1000-compound library. 19 The identified tool compounds were inhibitors of GSK-3 (x3), inhibitors of p38 MAP kinase (x2), agonists of α-adrenoceptor (x2), modulators of cyclin-dependent kinases (x2; Cdk2, Cdk1/Cdk2/Cdk5), modulators of other neurotransmitters (x3; histamine production, histamine receptor, 5-HT receptor), modulators of ion channels (x2; Na+-channels, L-type Ca2+-channels), other kinase inhibitors (x3; unselective, PKA/PKC/MLCK, PKA/CaM kinase III), a ROCK I+II inhibitor, a CNS convulsant, a Ca2+ ionophore, a RAR (α,β,γ) agonist, a PDE V inhibitor, and a Cdc25A phosphatase inhibitor. Five of those compounds were identified as hits in both of the two independent screens, and the top four hits generated screening results that were greater than three SDs above the mean values of the negative control data points ( Table 1 ). Follow-up studies confirmed a clear dose-dependent effect for one of the five compounds, which was a cdk2 modulator ( Fig. 5 ), with a significant positive effect in both screens.
Results of Screening
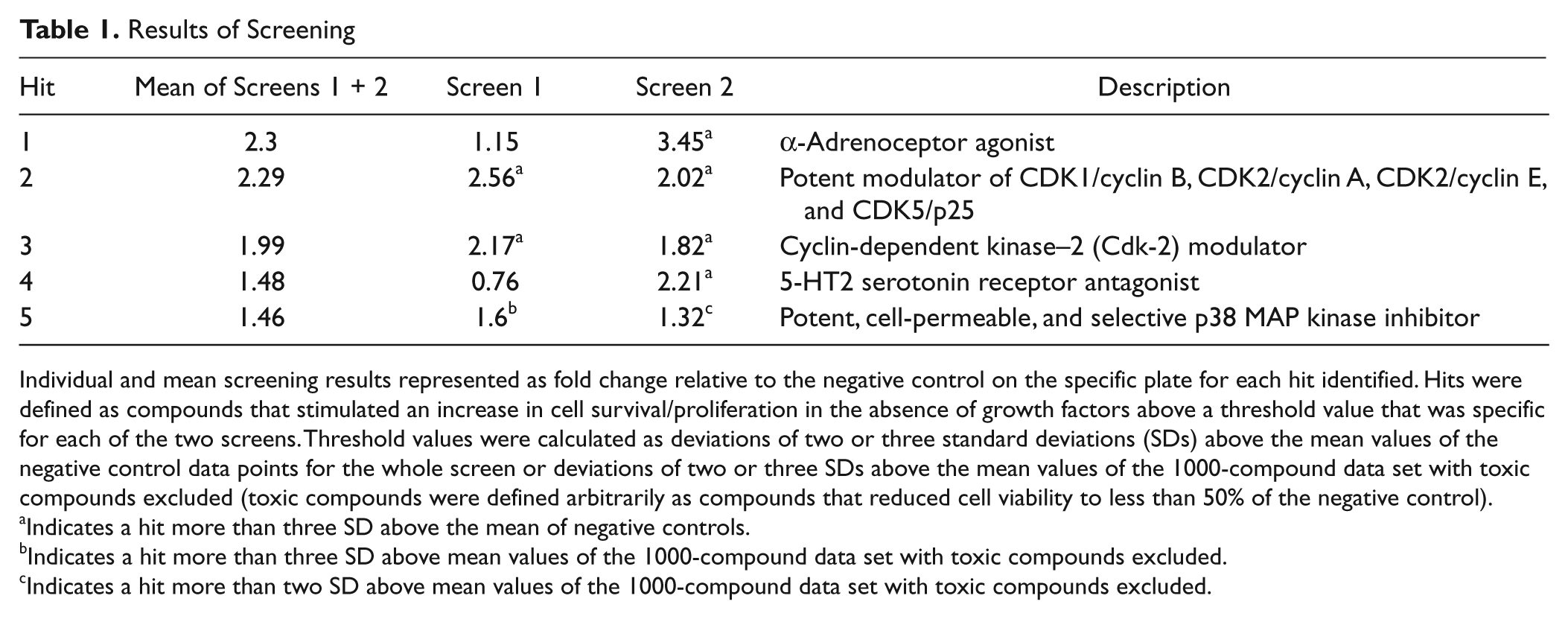
Individual and mean screening results represented as fold change relative to the negative control on the specific plate for each hit identified. Hits were defined as compounds that stimulated an increase in cell survival/proliferation in the absence of growth factors above a threshold value that was specific for each of the two screens. Threshold values were calculated as deviations of two or three standard deviations (SDs) above the mean values of the negative control data points for the whole screen or deviations of two or three SDs above the mean values of the 1000-compound data set with toxic compounds excluded (toxic compounds were defined arbitrarily as compounds that reduced cell viability to less than 50% of the negative control).
Indicates a hit more than three SD above the mean of negative controls.
Indicates a hit more than three SD above mean values of the 1000-compound data set with toxic compounds excluded.
Indicates a hit more than two SD above mean values of the 1000-compound data set with toxic compounds excluded.
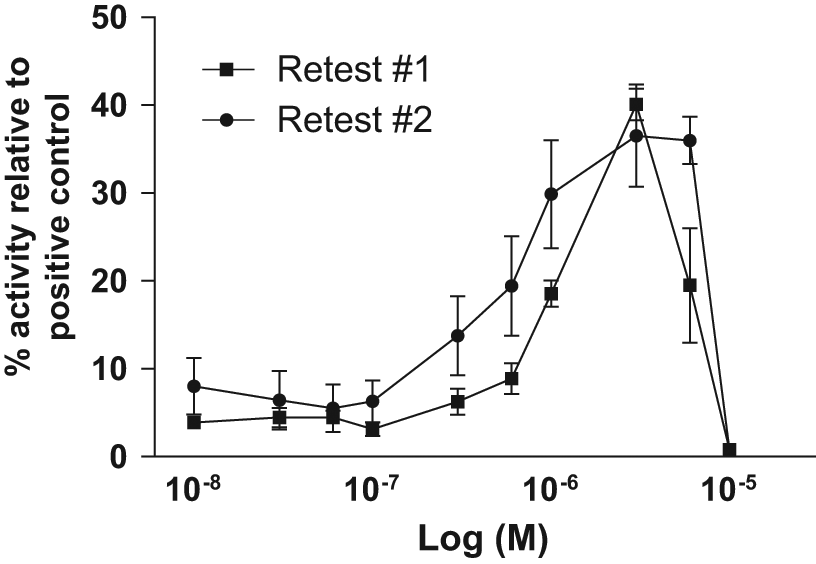
Dose-dependent response of compound 3. Using freshly supplied compound, hit confirmation was demonstrated with a dose-dependent effect when compound 3 ( Table 1 ) was tested on two independent occasions (retest 1 shown with squares, retest 2 shown with circles). The compound 3 dose-dependent effect was evaluated by plating 2000 neuroepithelial-like stem (lt-NES) cells per well in a 96-well plate format in the presence of increasing concentrations of compound and performing the adenosine triphosphate (ATP)–bioluminescence cell viability assay after 4 days of incubation (n = 3 wells per data point).
The physiochemical properties of the three identified NCEs make them suitable for future medicinal chemistry optimization, with molecular weight, LogP, H-bond donor/acceptor counts, and number of rotable bonds all within Lipinski’s parameters for drug likeness ( Table 2 ). The NCE with the highest level of activity identified at the screening concentration of 1 µM had a mean fold change of 1.27 relative to the negative control in the two independent screens (data not shown).
Physiochemical Properties of New Chemical Entity Hit Compounds
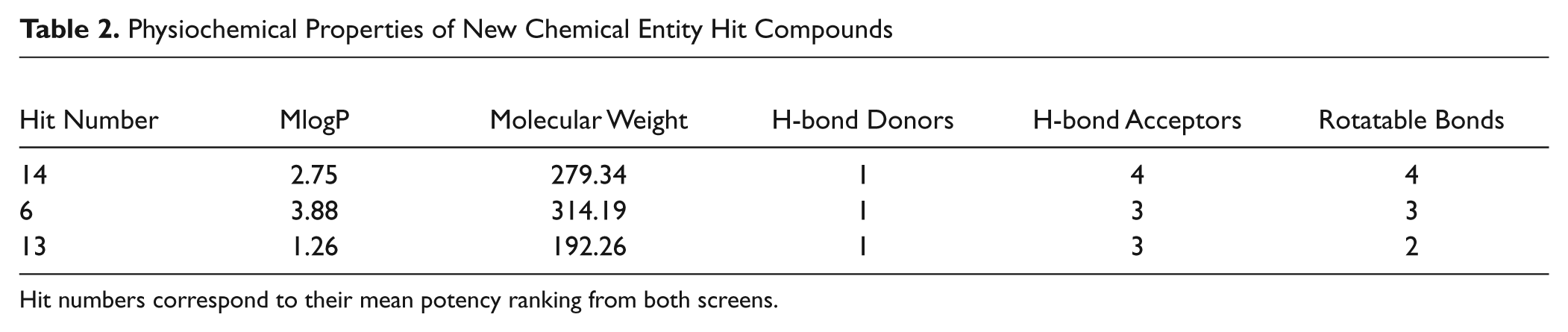
Hit numbers correspond to their mean potency ranking from both screens.
Discussion
The human iPS-derived lt-NES cells have demonstrated their utility in satisfying industry requirements for HTS in that they are readily expandable by robotic automation, they are amenable to cell-based assays in a microwell plate format, and proof of concept for a compound library screen has been demonstrated. In the latter, the chosen ATP-bioluminescence assay, for a proof-of-concept HTS-capable screening methodology, was shown to be both robust by Z′ factor screening metrics and predictive of the relevant functional cell biology by identifying several pathways that were previously reported to be involved in the control of NSC self-renewal.
To our knowledge, this is the first published example demonstrating robotic automation of a nonimmortalized human pluripotent stem cell–derived, stably propagating neural stem cell line. Our group previously reported the robotic automation of nonimmortalized and others the automation of immortalized human CNS tissue-derived NSCs.23,24 Our data demonstrate that robotic long-term automated culture, scale-up, and scale-out in HTS-ready microwell plates are feasible for human lt-NES cells on a scale sufficient to screen thousands of compounds per week. Although some drug discovery companies have previously been hesitant to adopt stem cell–based screening technologies due to the perceived difficulty of growing and maintaining fastidious stem cell lines in the appropriate undifferentiated format, the fact that our processes are readily transferable between platforms may encourage a change in that policy since many pharmaceutical companies already operate automated cell culture platforms to expand and maintain their cell lines for screening. Furthermore, automation protocols can be easily uploaded, making possible the straightforward transfer of cells and protocols between laboratories, thereby enabling the potential implementation of human neural stem cell–based screening activities in drug discovery labs that have no prior stem cell expertise.
Daily microscopic evaluation of the cell cultures for inspection of confluency currently constitutes a bottleneck in their automation. However, this could be resolved with the incorporation of an Incucyte (Essen BioScience, Ann Arbor, MI) into the CompacT SelecT. The automated cascade could be further improved by a recent manufacturer’s upgrade to the CompacT SelecT platform, which will enable the automated addition of small volumes that can be stored at 2 to 8 °C, negating the requirement for the static tube addition in our protocol; by incorporation of a centrifuge, which would greatly enhance the repertoire of serum-free stem cell cultures that could easily be automated; and by software enhancements increasing the flexibility of programming for stem cell automated maintenance. To fully automate this process to a scale capable of satisfying HTS requirements, it would be a necessity to further miniaturize the assay to a 384-well format, and further investigation would be required to validate whether an automated media change could be incorporated into the process after plating cells in a 384-well format. Furthermore, there would be the additional challenge of determining a suitable incubation time for the assay in a 384-well format given the increased risk of damaging evaporation as culture volumes are further reduced.
To date, only a few examples exist in the literature for assays and screens testing compounds for their influence on the proliferation of human NSCs. A study using immortalized human neural progenitors only identified inhibitors of proliferation, because the immortalization likely renders molecules and pathways that would normally potentiate self-renewal and proliferation ineffective. 25 Diamandis et al. 26 conducted a chemical genetic screen of similar size to ours for effects on the proliferation of mouse SVZ-derived neurosphere NSCs and as principal finding reported that proliferation was inhibited by perturbation of a number of neurotransmitter pathways. However, because the bulk of the cells in their neurosphere cultures were committed neural precursors and not NSCs, it is difficult to discriminate whether their identified hits acted on precursor cells rather than on NSCs. A more homogeneous stem cell population, as used in our current study, makes the results of such screens easier to interpret.
The 1000-compound screen we present here successfully identified 24 hit compounds that show an increase in survival or proliferation. In particular, hits from the tool compounds collection indicate that GSK-3 inhibition, cell cycle modulation, p38 MAP kinase inhibition, and α-adrenoceptor agonism may be modes of action that warrant further investigation, because multiple hits for each mode appear to confirm these as validated targets. That the assay hit results are highly germane to actual modulation of NSC biology is further corroborated by the fact that these modes of action were previously identified to influence NSC behavior in other assays or in vivo. Wnt3 overexpression, and therefore enhanced signaling akin to inhibition of GSK3, was sufficient to increase neurogenesis from adult hippocampal progenitors in vitro and in vivo, 27 and conditional deletion of GSK3 in mouse neural progenitors resulted in their massive hyperproliferation along the entire neuraxis. 28 It has been amply demonstrated that modulation of the cell cycle can influence both the rate and mode (symmetric vs. asymmetric) of NSC division, as reviewed by Salomoni and Calegari. 29 Androutsellis-Theotokis and coworkers 30 showed that inhibition of p38 MAP kinase, downstream of LIF receptor mediators, promoted self-renewal in NSCs at the expense of differentiation. This was independently confirmed by the identification of p38 as an NSC self-renewal modulator in a screen of a pharmacologically active compound library. 26 Last, stimulation of α1-adrenergic receptors was reported to enhance the proliferation of rodent NSCs in vitro. 31 There was also a substantial overlap of seven compounds from the 24 hits identified with human lt-NES cells in our hands, with a previous survival/proliferation screen of the same 1000-compound library on mouse adherent NSCs, 19 indicating that at least a proportion of hit compounds screened with rodent cells may translate to a human NSC-based model. Nevertheless, corroboration of pathways influencing proliferation and self-renewal occurred not only for previous in vitro and in vivo studies with rodent NSCs but also with the identification of a Rho-associated protein kinase I+II (ROCK) inhibitor in our screen, since the results of the only other published cell-based assay study for the same AF22 human lt-NES cell line identified three different ROCK inhibitors to promote their survival/self-renewal. This study, which screened a 160-compound small-molecule library for effects on proliferation, used a brightfield image label-free, high-content time-lapse assay system (Incucyte; Essen BioScience) that automatically measured cell confluence percentage over a 5-day period. 20
However, caution has to be exercised when interpreting the results from a homogeneous assay for proliferation, because NSCs are different from cancer and immortalized cell lines in the sense that they can undergo self-renewing symmetric divisions, generating more stem cells, or asymmetric divisions, generating basal, intermediate progenitors, capable of a few more rapid divisions, but finally leading to commitment and differentiation. 29 Although under standard expansion culture conditions, lt-NES cells divide symmetrically as a homogeneous self-renewing population, it is unclear whether this occurs once the cells are placed under assay conditions with small-molecule compounds promoting their proliferation. The short-term ATP-bioluminescence–based cell viability assay cannot discriminate between the two modes of NSC division, and to determine whether a pro-proliferative hit compound from the homogeneous screen would indeed expand the NSC pool, a validated follow-on phenotypic assay for self-renewal will be essential. We developed such a self-renewal assay for NSCs on a high-content screening platform (IN Cell 2000; GE Healthcare, Piscataway, NJ), which quantifies immunostaining percentage and intensity of the NSC multipotency maintaining Sox2 master gene. This assay was validated by detecting a significant reduction in Sox2 staining intensity of NSCs, when either EGF and FGF2 were withdrawn for 72 h or the self-renewal disrupting and differentiation-inducing Notch-signaling inhibitor DAPT was applied to the cultures in the presence of full growth factor conditions for 72 h (data not shown).
Another important application of the ATP-bioluminescence–based cell viability assay that we present here lies in the potential to discover reagents that will reduce the cost of employing a stem cell technology platform for HTS. Although improved prediction of efficacy and safety with human stem cell–based systems over recombinant and cancer cell lines is generally accepted by industry, a key challenge to their wider utilization for primary HTS is the current practice of maintenance and modulation of stem cell populations, which is achieved through the use of complex and expensive cocktails of recombinant protein reagents and, in the case of adherent cultures, with equally costly purified extracellular matrix proteins. This runs contrary to the commercial imperative to keep the cost per well/screened compound as minimal as possible for primary HTS. The use of small molecules and polymer chemistry matrices should be able to substitute many components of these high-cost reagents, significantly reducing the associated cost of goods and providing a cheap, versatile, and tunable tool kit of reagents to enable cost-effective primary and safety screening for academia and the pharmaceutical industry.
In this study, we demonstrate that protocols devised to derive human long-term stably proliferative neuroepithelial-like stem cells (human lt-NES) from induced pluripotent stem (iPS) cells are amenable to automation, thereby enabling the in vitro comparison of the properties of NSCs from both normal and disease-specific phenotypes. Stably proliferating, homogeneous, and multipotent lt-NES cells derived from iPS cells have tremendous potential to be incorporated into the development of high-throughput platforms for the screening of small molecules that specifically target cells with diseased phenotypes alongside normal controls. Utilization of iPS-derived lt-NES cells for screening offers several advantages over run-through protocols that rely on neural differentiation of the iPS source cells themselves. First, this approach reduces the risk of contaminating nonneural cell types and residual pluripotent cells following differentiation. Second, the iPS-derived lt-NES cells provide a more uniform differentiation into neural cell types than would likely be generated if a differentiation protocol was initiated each time from iPS cells. Although the lt-NES cells can be screened for modulators of NSC proliferation and differentiation, as reported in this study, or for drugs and environmental agents that could cause developmental neurotoxicity, mature neurons derived of these cells should prove useful in primary screens for neuroprotection and in drug safety screens for neurotoxicity and synaptic dysfunction. Another translational application of lt-NES, demonstrated in a recent study, 20 is as an ideal cell population for a counterscreen of small molecules that will selectively kill or inhibit the growth of brain tumor stem cells but have no detrimental effect on normal NSCs resident in the neurogenic niches of the brain.
Footnotes
Acknowledgements
We thank Dr Ann Tsukamoto for expert advice and Sarita Maman for assistance with assay development.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: LT-NES is a registered trademark of LIFE&BRAIN GmbH. A. S. is a medical research council professor. This work was partially supported by the European Commission Research Framework projects NEUROscreen (LSHB-CT-2007/037766) and SCR&Tox (HEALTH-F5-2010-266753) and the state of North Rhine Westphalia (BIO.NRW; Project StemCellFactory).