
Abstract
Non–adenosine triphosphate (ATP) competitive, allosteric inhibitors provide a promising avenue to develop highly selective small-molecule kinase inhibitors. Although this class of compounds is growing, detection of such inhibitors can be challenging as standard kinase activity assays preferentially detect compounds that bind to active kinases in an ATP competitive manner. We have previously described a time-resolved fluorescence resonance energy transfer (TR-FRET)–based kinase binding assay using the competitive displacement of ATP competitive active site fluorescent probes (“tracers”). Although this format has gained acceptance, published data with this and related formats are almost entirely without examples of non-ATP competitive compounds. Thus, this study addresses whether this format is useful for non-ATP competitive inhibitors. To this end, 15 commercially available non-ATP competitive inhibitors were tested for their ability to displace ATP competitive probes. Despite the diversity of both compound structures and their respective targets, 14 of the 15 compounds displaced the tracers with IC50 values comparable to literature values. We conclude that such binding assays are well suited for the study of non-ATP competitive inhibitors. In addition, we demonstrate that allosteric inhibitors of BCR-Abl and MEK bind preferentially to the nonphosphorylated (i.e., inactive) form of the kinase, indicating that binding assays may be a preferred format in some cases.
Introduction
There is a continued need to develop selective, high-affinity small-molecule protein kinase inhibitors. Although most kinase inhibitors described to date are adenosine triphosphate (ATP) competitive, there is increasing interest in the development of kinase inhibitors that are non-ATP competitive and bind outside of the ATP binding site, commonly referred to as allosteric inhibitors. Such compounds may provide higher specificity and improved therapeutic efficacy. Their mechanisms of action are varied, with some being competitive with the target’s protein substrates, some binding to the target of interest and inhibiting its interaction with an upstream activating kinase, and others binding to a site outside the ATP pocket and causing conformational changes at a second, distal site, inducing an inactive conformation.
Promising data suggest that the use of an allosteric kinase inhibitor in combination with an ATP competitive inhibitor of the same target can improve efficacy and may help to overcome resistance to either compound alone.1,2 GNF-2 and GNF-5 are unique Bcr-Abl inhibitors that bind to a myristoyl binding pocket located on the carboxyl terminus of the Abl kinase domain.3,4 Binding of these inhibitors to this site causes perturbations in residues within the ATP-binding site, 5 and when used in combination with either imatinib or nilotinib, the T315I Bcr-Abl mutant, resistant to either compound alone, is inhibited. 2 Structural data suggest that non-ATP competitive inhibitors of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK) also bind to and stabilize an inactive conformation of the protein where conformational changes of the activation loop as well as shifts in the location of the C-helix are observed. 6 Screening efforts to identify and characterize allosteric kinase inhibitors have been hampered by the fact that most biochemical kinase activity assays preferentially detect compounds that bind to active kinases in an ATP competitive manner. Given the structural perturbations observed at the active site in the presence of several of these allosteric inhibitors, we questioned whether these perturbations could be detected by displacement of active site probes. We have previously demonstrated the utility of a binding assay based on the displacement of small-molecule ATP competitive kinase inhibitor scaffolds that are conjugated to a fluorescent dye. Binding of this probe (referred to as a “tracer”) to the kinase of interest is detected using a europium-labeled antitag antibody, which is bound to the kinase of interest at a glutathione S-transferase (GST) or polyhistidine-tagged site. When both tracer and antibody are bound, there is a high time-resolved fluorescence resonance energy transfer (TR-FRET) signal. When the tracer is displaced by a kinase inhibitor, there is a loss of the TR-FRET signal. 7 This method is particularly useful when examining compound binding to nonactivated (e.g., nonphosphorylated) kinase states. However, the question remains as to whether this format can be broadly used to detect and characterize non-ATP competitive inhibitors, despite being based on ATP competitive tracers. This study was designed to address this question.
Materials and Methods
Binding and activity assays were performed as previously described 7 in 384-well low-volume plates (Corning part #3673 or #3676; Corning, Inc., Corning, NY) at room temperature in kinase buffer A, which consists of 50 mM HEPES (pH 7.5), 0.01% Brij-35, 10 mM MgCl2, and 1 mM EGTA with the exception of the Abl assay, which was performed in the same buffer without addition of Brij-35 and the MEK assays, which in some cases were performed in the presence of subsaturating concentrations of ATP. Kinase buffer A, europium (Eu)– and terbium (Tb)–labeled antibodies, streptavidin, Alexa Fluor dyes, kinases, and kinase tracers were from Life Technologies (Carlsbad, CA). Sequences for the kinases used in this study match the GenBank accession numbers reported in the supplementary information. Kinase inhibitors were obtained from Alexis Biochemicals (San Diego, CA), ChemieTek (Indianapolis, IN), EMD Biosciences (San Diego, CA), Fermentek (Jerusalem, Israel), LC Laboratories (Woburn, MA), Life Technologies, Selleck Chem (Houston, TX), Sigma Aldrich (St. Louis, MO), Stemgent (Cambridge, MA), and Tocris (Minneapolis, MN). Specific assay conditions were as follows with all kinases at 5 nM unless noted and antibody and Eu-streptavidin at 2 nM:
IKKα (CHUK): 30 nM Kinase Tracer 236, biotin anti-His, Eu-streptavidin
IKKβ (IKBKB): 80 nM Kinase Tracer 236, Eu–anti-GST
IKKϵ (IKBKE): 1 nM kinase, 1 nM Kinase Tracer 236, Eu–anti-GST
Abl: 15 nM kinase (based on a 50% purity estimate), 125 nM Kinase Tracer 1710, biotin–anti-His, Eu-streptavidin
MEK1 (MAP2K1), inactive: 50 nM Kinase Tracer 236, biotin anti-His, Eu-streptavidin
In vitro phosphorylated MEK1: 7 nM Tracer 236, biotin anti-His, Eu-streptavidin
AKT1: 100 nM Kinase Tracer 419, biotin–anti-His, Tb-streptavidin
AKT2: 50 nM Kinase Tracer 419, biotin–anti-His, Tb-streptavidin
AKT3: 40 nM Kinase Tracer 419, biotin–anti-His, Tb-streptavidin
GSK3α and GSK3β: 10 nM Kinase Tracer 236, Eu–anti-His
p38α (MAPK14): 1 nM kinase, 30 nM Kinase Tracer 199, Eu–anti-GST
Binding assays were performed with tracer concentrations near or below the experimentally determined Kd (App) concentration as previously described,
7
and an example of this analysis can be found in
Assay plates were read after incubation for 1 h at room temperature or as specified with a BMG LABTECH PHERAstar (BMG LABTECH GmbH, Ortenberg, Germany) using standard Eu-based TR-FRET settings with excitation at 340 nm and emission monitored at 615 nm (donor) and 665 nm (acceptor) or Tb-based settings with excitation at 340 nm and emission monitored at 490 nm (donor) and 520 nm (acceptor). Emission intensities were measured over a 200-µs window following a 100-µs postexcitation delay. Data are presented either as a ratio of raw acceptor/donor intensities or normalized relative to wells containing fully bound or fully competed tracer (relative percent tracer bound).
In Vitro Phosphorylation of MEK1 (MAP2K1)
Purified wild-type MEK1 (MAP2K1) expressed in insect cells was phosphorylated in vitro by incubation with an oncogenic BRAF mutant V600E (formerly referred to as V599E)as follows; 500 nM MEK1 was incubated with 10 nM BRAF V600E in kinase buffer A containing 200 µM ATP for 1 h at room temperature. The ATP was removed from the reactions by applying the sample to an NAP 10 column (GE Healthcare, Piscataway, NJ) and eluting with kinase buffer A.
MEK1 Cascade and Direct Activity Assays
A triple-cascade assay was performed under optimized conditions by Life Technologies’ SelectScreen Kinase Profiling Service. Briefly, inhibitor titrations were tested in 10-µL kinase reactions containing 0.04 to 0.15 ng BRAF, 10 ng inactive MAP2K1 (MEK1), 100 ng inactive MAPK1 (ERK2), and 2 µM Ser/Thr 03 peptide in 50 mM HEPES (pH 7.5), 0.01% Brij-35, 10 mM MgCl2, and 1 mM EGTA and were initiated with 100 µM ATP, allowed to proceed for 1 h, and then developed as previously described. 8
A direct MEK1 assay was developed using GFP-ERK2 and a terbium-labeled anti-ERK2 [pThr185/pTyr187] antibody. Inhibitor titrations were tested in 10-µL kinase reactions containing 1.2 µg/mL MEK1 and 100 nM GFP-ERK2 in kinase buffer A. Reactions were initiated with 8 µM ATP and allowed to proceed for 1 h. Reactions were stopped with 10 mM EDTA and phosphorylation of the ERK2 detected by 2 nM anti-ERK2 [pThr185/pTyr187] antibody.
Measurement of Inhibitor Binding to Active or Nonactivated Abl
Full-length His-tagged human recombinant Abl (isoform 1a, which does not contain the N-terminal myristoylation site) was expressed in baculovirus-infected insect cells and purified in the absence of detergent to approximately 50% purity. This preparation was treated with YOP protein tyrosine phosphatase as follows; 0.4 mg/mL of the sample was incubated at room temperature for 1 h with 3.6 units of YOP protein tyrosine phosphatase (PTP) per µL in a total volume of 110 µL in 50 mM HEPES (pH 7.5), 10 mM MgCl2, and 1 mM EGTA. Binding assay conditions were optimized for both activated and phosphatase-treated (nonactivated) Abl. Assays were performed in 50 mM HEPES (pH 7.5), 10 mM MgCl2, and 1 mM EGTA and were incubated 2 h prior to data acquisition. To determine the efficacy of the phosphatase treatment, a homogeneous TR-FRET assay was employed as follows; 10 nM kinase was incubated with 5 nM Eu-labeled PY-20 antibody and 5 nM Alexa Fluor 647–labeled anti-His antibody in kinase buffer A in a final reaction volume of 15 µL. Reactions were incubated 1 h and then read as described for the kinase binding assay above. Emission ratios were compared between YOP-treated, mock-treated, and a no-kinase control.
Results and Discussion
Analysis of Non-ATP Competitive MEK Inhibitors in the Presence and Absence of ATP: Binding Affinities Are Phosphorylation-State Dependent
Ras/Raf/MEK/ERK/MAPK signaling pathways are critical for cellular growth and differentiation, and loss of regulation of these pathways leads to proliferative and inflammatory diseases. Because structural perturbations at the active site have been observed for allosteric compounds binding to MEK,
6
we tested these compounds in a competitive displacement binding assay. We developed assays for both the active and nonactive forms of the kinase. In addition, because three of these allosteric MEK inhibitors (AZD6244, PD0325901, and PD184352 [CI-1040]) have been shown to bind in an uncompetitive manner with respect to ATP—namely, that they bind with higher affinity in the presence of ATP9–11—we developed methods to test the displacement of the active site probe in the presence of subsaturating concentrations of ATP. After optimization of assay conditions in the absence of nucleotide, we titrated both ATP and adenosine diphosphate (ADP) to determine IC50 values for nonactivated, nonphosphorylated MEK1 (npMEK1) and activated, in vitro phosphorylated MEK1 (pMEK1). The affinity observed for ATP versus ADP was similar for each kinase variant tested (1.4 µM for ATP and 1.2 µM for ADP for npMEK1 and 3.1 µM for each nucleotide for pMEK1). An example of an ATP titration curve is illustrated in
To explore changes in affinity in the presence of nucleotide, inhibitor titrations were performed in the presence or absence of subsaturating concentrations of ATP in the binding assay. We chose 0.5 µM ATP for npMEK1 and 2 µM for pMEK1. These values were selected from titration curves of ATP to ensure a sufficient assay window for the inhibitor studies (
The ATP uncompetitive MEK inhibitors demonstrate dramatic shifts in IC50 values upon the addition of subsaturating concentrations of ATP ( Fig. 1 and Table 1 ). Each of these compounds binds preferentially to npMEK1 in the presence of ATP with the largest shifts observed for AZD6244. There is a greater than 900-fold difference in the IC50 value observed for the phosphorylated MEK1 in the absence of ATP (IC50 value of 7400 nM) versus the nonphosphorylated form in the presence of ATP (IC50 value of 8 nM). Each of the ATP uncompetitive inhibitors fully displaces the tracer from npMEK1 in the presence of ATP; however, PD184352 exhibits partial displacement of the tracer in the absence of ATP, with maximal displacement of 60% from the phosphorylated form in the absence of ATP.
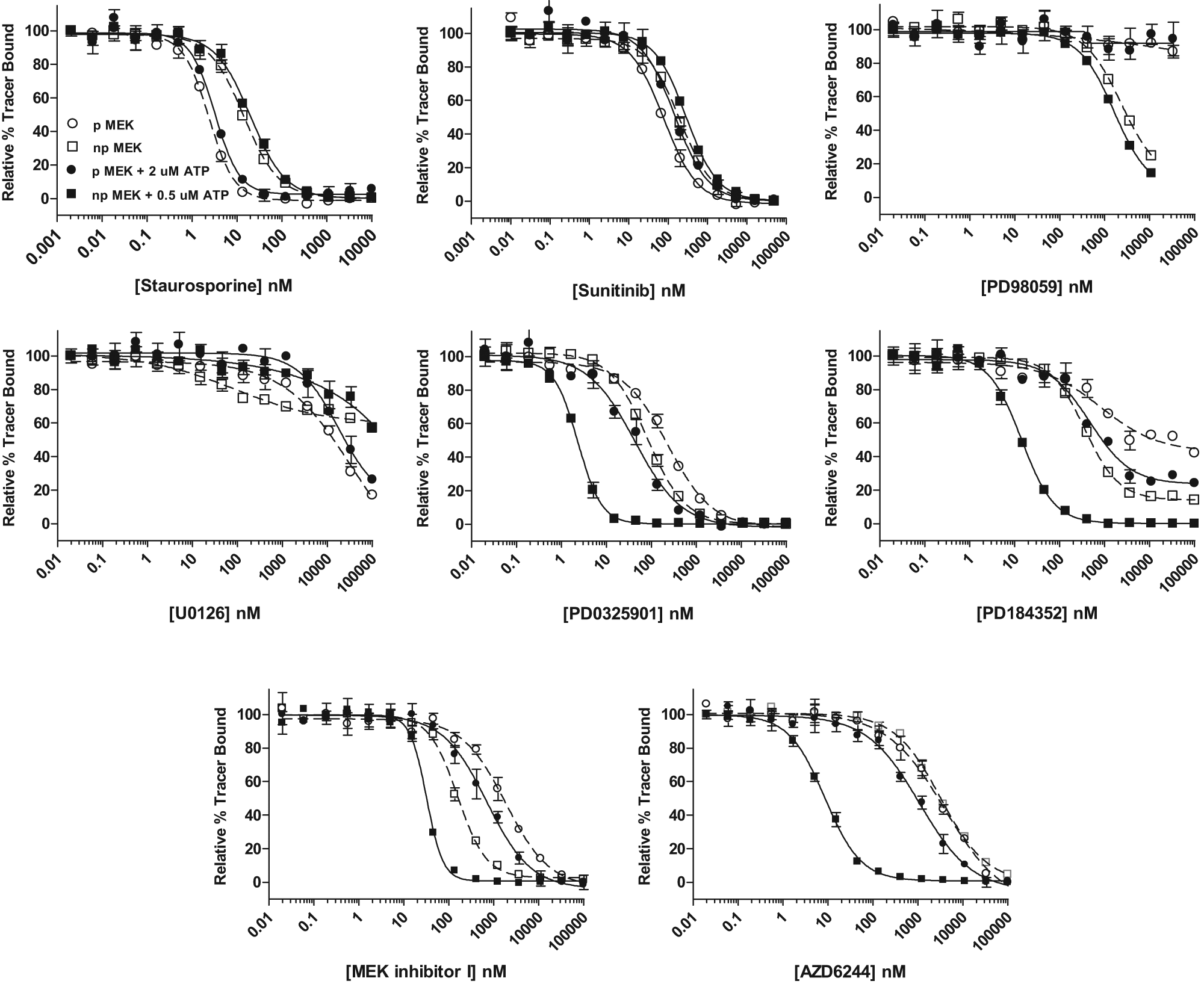
Analysis of diverse mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK) inhibitors binding to nonphosphorylated MEK1 (npMEK1) and in vitro phosphorylated MEK1 (pMEK1). Inhibitor binding to nonactivated, nonphosphorylated MEK1 in the absence (□) or presence of 0.5 µM adenosine triphosphate (ATP; ■) or phosphorylated MEK1 in the absence (○) or presence (•) of 2 µM ATP. One of three independent experiments is shown, with error bars representing the standard deviation of three replicates.
Summary of MEK1 Inhibitor Data
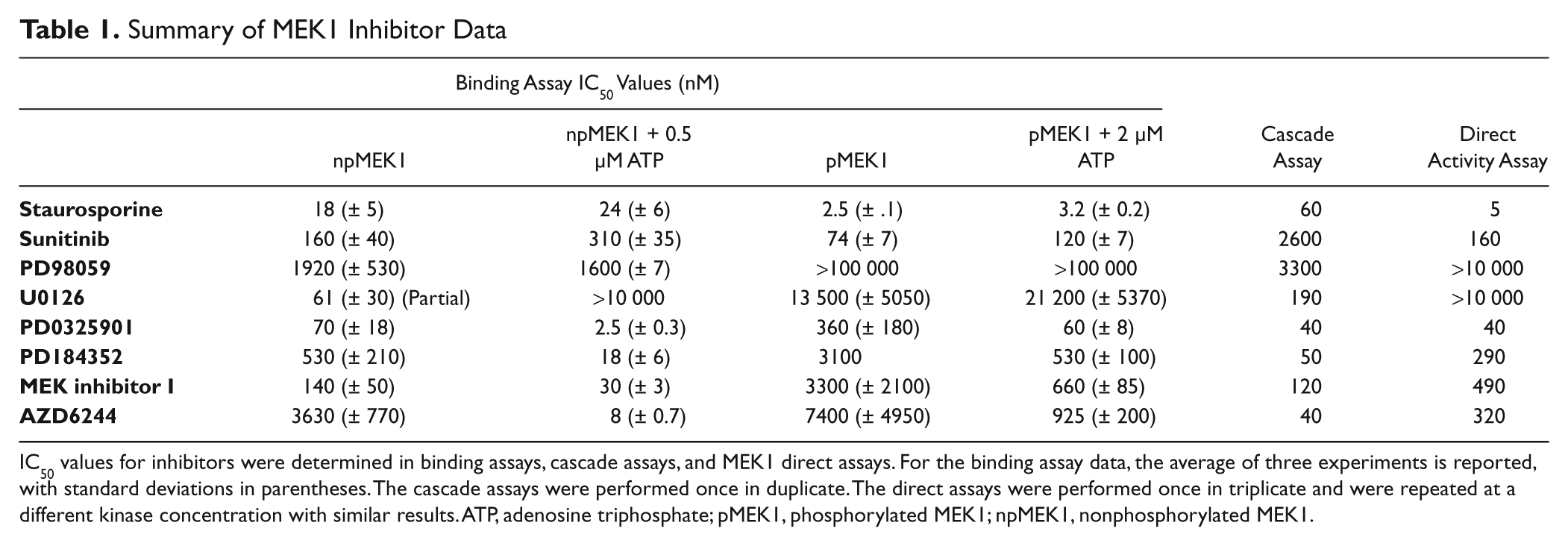
IC50 values for inhibitors were determined in binding assays, cascade assays, and MEK1 direct assays. For the binding assay data, the average of three experiments is reported, with standard deviations in parentheses. The cascade assays were performed once in duplicate. The direct assays were performed once in triplicate and were repeated at a different kinase concentration with similar results. ATP, adenosine triphosphate; pMEK1, phosphorylated MEK1; npMEK1, nonphosphorylated MEK1.
To more fully understand these data, activity-based assays were performed for comparison. A triple-cascade assay, BRAF-MEK1-ERK2, and a direct assay, assessing the phosphorylation of GFP-ERK2 by MEK1, were employed ( Table 1 ). As expected, the direct assay demonstrated very similar IC50 values for the ATP competitive inhibitors, staurosporine and sunitinib, as were observed for the binding assay (5 and 160 nM, respectively). The cascade assay demonstrated a right-shifted IC50 value for both of these inhibitors as expected due to the assay being performed above the MEK1 ATP Km at 100 µM ATP. Consistent with binding to the nonactivated, nonphosphorylated form of MEK1 and literature values, the IC50 value for PD98059 was 3.3 µM in the cascade assay, and it did not show inhibition in the direct assay. Likewise, U0126 fully inhibited the cascade assay with an observed IC50 value of 190 nM but only showed inhibition of 25% in the direct activity assay at 10 µM compound. The IC50 values observed for the ATP uncompetitive inhibitors in the direct activity assay were higher than those observed for the binding assay for the npMEK1 but were very similar to the binding assay results for the phosphorylated form of MEK1 and with the cascade assay generally demonstrating lower IC50 values, confirming that these inhibitors bind preferentially to the nonphosphorylated form of MEK1.
Analysis of GNF-2 Binding to Abl1
N-terminal myristoylation of Abl plays a critical role in the regulation of this kinase as binding of the myristoylated N-terminus to a myristic acid binding pocket in the C-lobe of the kinase domain is believed to induce an inactive kinase conformation. GNF-2 is a non-ATP competitive inhibitor of the constitutively active Bcr-Abl oncoprotein and Abl kinase. GNF-2 has been shown to bind to this same myristoyl binding pocket, 4 thereby mimicking the natural downregulation of kinase activity. Initial reports suggested that GNF-2 was not capable of inhibiting Abl in vitro kinase activity assays, but it was recently demonstrated that the nonionic detergent Brij-35 rendered GNF-2 an ineffective inhibitor, and in the absence of the detergent, inhibition was observed. 4 To detect if the active site changes induced by GNF-2 could be detected by displacement of an active site probe, we purified nonmyristylated Abl and developed displacement assays in the absence of detergent. Because we had previously demonstrated that dephosphorylated Abl had different binding profiles than active (phosphorylated) Abl for several type II kinase inhibitors, 7 we also treated this detergent-free preparation with YOP phosphatase and optimized binding assay conditions for this preparation. As a control, we tested the displacement of the tracer with the ATP competitive inhibitor VX-680 and observed very similar IC50 values between the untreated and phosphatase-treated kinase ( Fig. 2A ). As expected, dephosphorylated Abl exhibited a nearly 10-fold increased affinity for imatinib (60 nM untreated vs. 6.5 nM phosphatase treated; Fig. 2C ). With this detergent-free preparation, we observed higher affinity binding to the untreated sample than has been observed with active Abl purified in the presence of detergent, 7 suggesting that the preparation contains a higher proportion of nonactivated Abl. Displacement of the tracer by GNF-2 was also dramatically affected by phosphatase treatment. Incubation of GNF-2 with untreated Abl resulted in maximal displacement of the tracer of 35% at 10 µM GNF-2 with a very shallow curve (Hill slope of 0.5) and an IC50 value of 350 nM. Treatment with phosphatase resulted in maximal displacement of the tracer of 55% to 65% (varying with experiment), a Hill slope of 1, and an IC50 value of 110 nM ( Fig. 2B ). To verify that the YOP treatment was dephosphorylating the kinase, we developed a homogeneous TR-FRET method to monitor the dephosphorylation of tyrosine residues. YOP- or mock-treated kinase was incubated with Eu-labeled anti-phosphotyrosine antibody and Alexa Fluor 647–labeled anti-His antibody. The emission ratio observed in the mock-treated sample was 6.1-fold higher than that observed for the no-kinase control sample ( Fig. 2D ), whereas the YOP-treated emission ration was only 1.3-fold higher than that observed for the no-kinase control sample, demonstrating that the YOP phosphatase was effectively dephosphorylating the kinase on tyrosine residues.
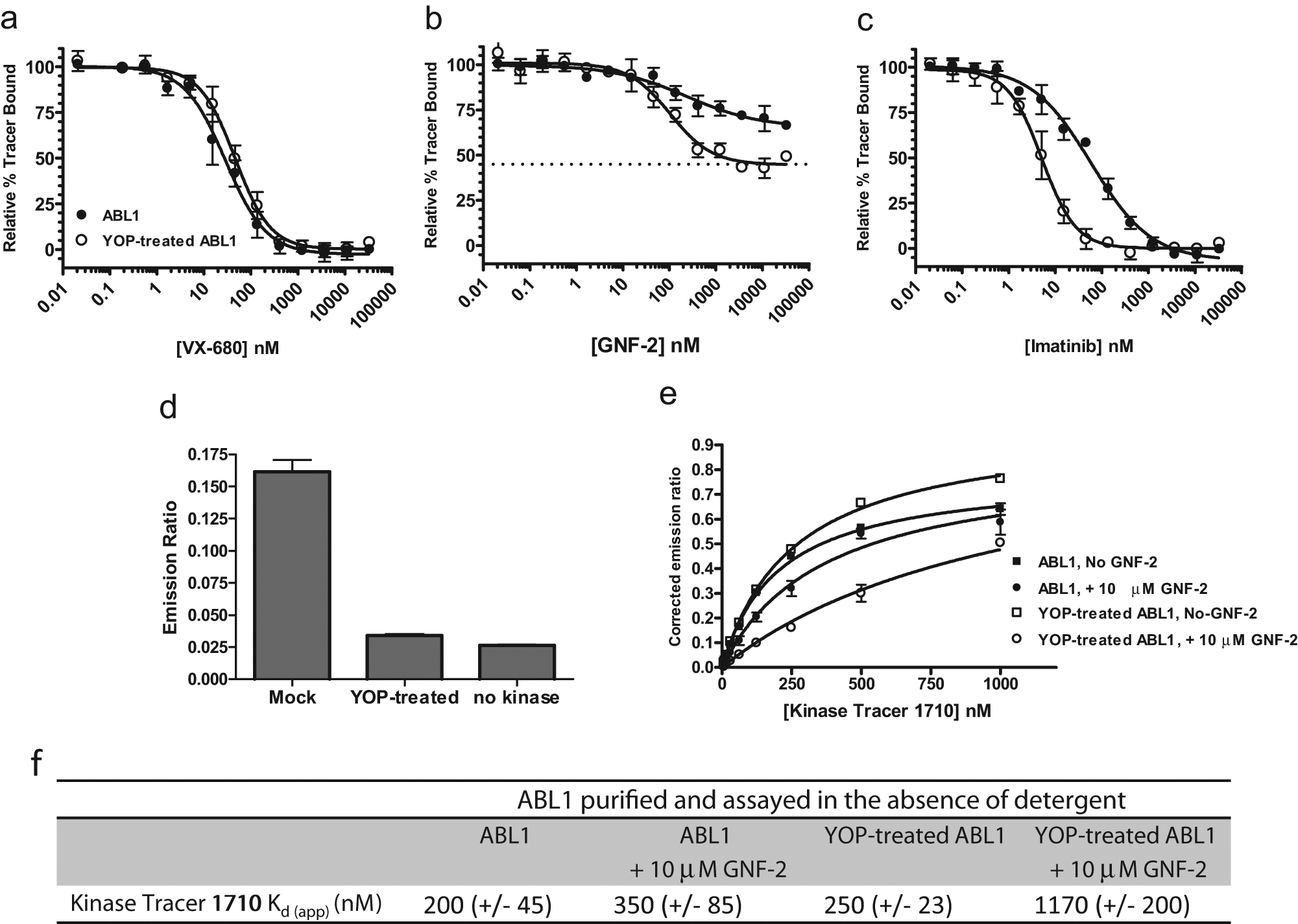
GNF-2 binds preferentially to nonactivated Abl1. Binding assays for nonmyristoylated Abl1 were optimized in the absence of detergent. Inhibitor titrations were performed with nontreated (•) and phosphatase-treated (○) samples (
The observation that binding of GNF-2 resulted in partial and not complete displacement of the tracer is notable. Because this assay format detects tracer binding and displacement to a variety of kinase conformations, it is possible that there is a population of the kinase that is not capable of binding GNF-2 and therefore is resistant to tracer displacement by this inhibitor. Alternatively, this could be the result of the inhibitor altering the affinity of the tracer to a lesser degree than that observed for other allosteric inhibitors where full tracer displacement is observed. Using this format, we cannot fully distinguish between these two possibilities, but we can test whether the kinase had lower affinity to the tracer in the presence of saturating amounts of the compound. To do so, we tested tracer affinity in untreated or YOP phosphatase-treated Abl in the presence or absence of 10 µM GNF-2. By this method, we detected a significant alteration in the affinity of the tracer for the YOP-treated kinase in the presence of GNF-2 ( Fig. 2E , F ). The affinity for Kinase Tracer 1710 was reduced in YOP-treated samples from 250 ± 23 nM in the absence of GNF-2 to 1170 ± 200 nM in the presence of 10 µM GNF-2. Statistical comparison of the data sets from three independent experiments generated a p value of 0.013, suggesting they are statistically different. Changes in the affinity of the tracer in non-YOP-treated samples were not statistically significant. This suggests that binding of GNF-2 to the C-lobe of the kinase domain does result in alteration of the active site sufficient to decrease tracer affinity, which is consistent with results obtained by hydrogen-exchange mass spectrometry analysis2,13 but does not rule out that there is a population of the kinase present that does not bind to GNF-2. When Abl is purified and assayed in the presence of nonionic detergents (purified with NP-40 present and assayed in the presence Brij-35), the ability of GNF-2 to displace the tracer is attenuated (data not shown). This supports the hypothesis proposed by Choi et al 4 that Brij-35 may occupy the myristate binding pocket and preclude binding of GNF-2 to the kinase.
Detection of Diverse Non-ATP Competitive Inhibitors
In the case of both the MEK1 inhibitors and GNF-2, there were previous structural data suggesting perturbation of the active site by these compounds. To test whether other, often less characterized non-ATP competitive kinase inhibitors would cause displacement of the active site tracers, we tested a comprehensive collection of commercially available non-ATP competitive inhibitors against their primary kinase targets in this competitive displacement binding assay. This diverse group of compounds included the plekstrin homology (PH) domain-dependent AKT inhibitors MK-2206 and AKT inhibitor VIII (AKTi 1/2) (supplemental references S1 and S2); three GSK3 inhibitors (supplemental reference S3); two p38α inhibitors, including JX401 and the substrate-specific inhibitor MK2a (supplemental references S4–S6); and the IKKβ inhibitor BMS-345541 (supplemental reference S7). Each of these inhibitors effectively displaced the tracer from the kinase with IC50 values that correlated well with literature values with the exception of the substrate-specific p38 inhibitor MK2a ( Table 2 ). The MK2a inhibitor specifically competes for MK2a binding to p38α but does not inhibit phosphorylation of MBP or ATF2 substrates (supplemental references S3 and S4), and therefore the active site remains catalytically competent and capable of binding the active site probe. Unlike the observation for U0126 and GNF-2, this set of inhibitors fully displaced the tracer from the kinase.
Detection of Diverse Allosteric Kinase Inhibitors by the Kinase Binding Assay
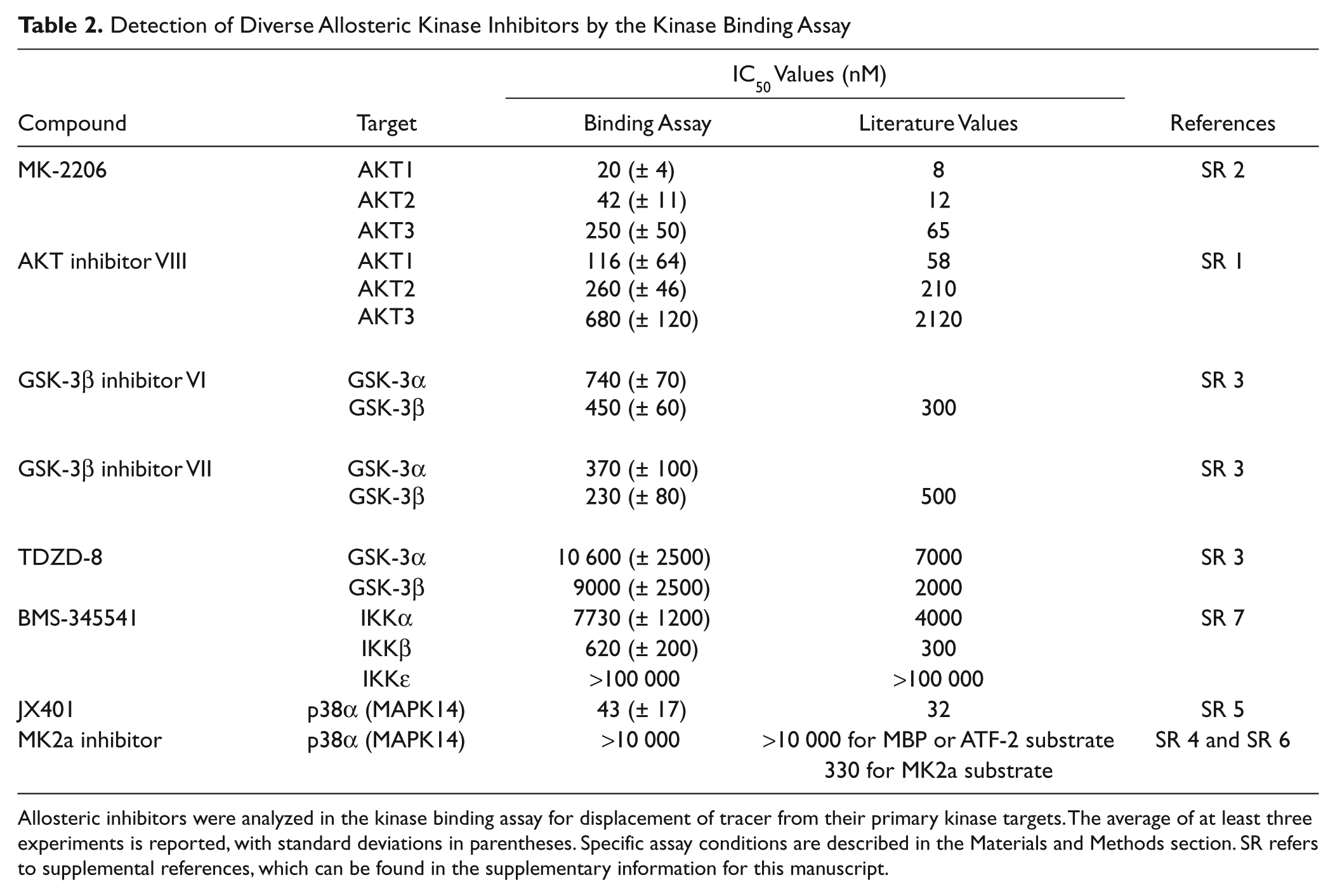
Allosteric inhibitors were analyzed in the kinase binding assay for displacement of tracer from their primary kinase targets. The average of at least three experiments is reported, with standard deviations in parentheses. Specific assay conditions are described in the Materials and Methods section. SR refers to supplemental references, which can be found in the supplementary information for this manuscript.
These data demonstrate that many of the diverse class of non-ATP competitive kinase inhibitors perturb the kinase active site as observed by altered affinity for and displacement of an active site probe. In general, ATP uncompetitive or ATP noncompetitive kinase inhibitors have been referred to as allosteric inhibitors, but these data elucidate a mechanistic difference between those that are protein substrate inhibitors and those that perturb the catalytic site from a distal binding location. One explanation for displacement of the active site probe is that the tracer and inhibitor occupy adjacent binding sites, and although the tracer is ATP competitive and can be fully displaced by ATP, it occupies a binding site larger than ATP alone and therefore is displaced upon binding of the allosteric inhibitor at the adjacent binding site. This remains a possibility for the non-ATP competitive MEK inhibitors, which do occupy a binding site adjacent to the ATP binding site. More difficult to explain, however, is the displacement of the tracer from AKT by the PH domain-dependent allosteric AKT inhibitors VIII (Akti-1/2) and MK-2206 and the myristate binding pocket inhibitor of Abl, GNF-2. It is not enough to hold the kinase in an inactive state akin to a dephosphorylated kinase, as in most cases that we have tested (and demonstrated here for MEK1 and ABL1), the tracers do bind to the nonactivated (nonphosphorylated) kinases. Thus, these inhibitors must induce a kinase state with a conformation such that the affinity of the tracer is dramatically lowered, resulting in dose-dependent displacement of the tracer. These states might resemble specific conformations that have been previously described such as one in which the C-helix is positioned incorrectly relative to the N-terminal lobe of the catalytic domain or one in which the hydrophobic spine described for many active kinases is misaligned.14,15
Unique challenges might arise in screening for allosteric kinase inhibitors. Screening of full-length proteins, rather than truncated catalytic domains, may be preferable and enable detection of compounds with novel binding modes. It is important that the proteins being screened must be able to sample their physiologically relevant conformations, and analyzing kinases in both their activated and nonactivated states might provide additional insights. This study validates competitive binding assays as an approach to the discovery and development of allosteric inhibitors that can be used with a broad sampling of the kinome in a format amenable to both screening and secondary analysis.
Footnotes
Acknowledgements
We thank Randy Hoffman and the SelectScreen Biochemical Kinase Assay group for performing the BRAF cascade assays, Brooke Richardson and Scott Brown for technical assistance with the MEK direct activity assay, and Kurt Vogel for critical review of this manuscript.
Notes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The authors are employed by Life Technologies Corporation.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article. Life Technologies Corporation funded this work.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.