
Abstract
Primary neurons in culture are considered to be a highly relevant model in the study of neuronal development and activity. They can be cultivated and differentiated in vitro but are difficult to transfect using conventional methods. To address this problem, a capillary electroporation system called Cellaxess Elektra was developed for efficient and reproducible transfection of primary cortical and hippocampal neurons without significant impact on cell morphology and viability. The cells are transfected in any stage of differentiation and development, directly in cell culture plates. Genetic material is delivered in situ to as many as 384 samples at a time, which enables both high-throughput and high-quality screening for hard-to-transfect primary cells, meaning that gene function can be studied on a genome-wide scale in cells previously inaccessible to genetic manipulation.
Introduction
In vitro models provide important tools for studying molecular and biochemical mechanisms involved in toxicity, cellular differentiation, and regulation of development. The utility of in vitro models is directly related to accurate representation of the in vivo system they are designed to mimic. 1 Because of availability and convenience, immortalized cell lines are often employed instead of primary cells. In the case of neuronal studies, few cell lines can serve as accurate in vitro models of neurons. Although often derived from neuronal cells, these lines typically consist of immortalized or cancerous cells that divide uncontrollably, suggesting their pattern of gene and/or protein expression may be significantly different from terminally differentiated and functioning neurons. 2 In terms of cellular composition and complexity, dissociated primary neuronal cell cultures more closely resemble in vivo biology than immortalized cell lines and represent a model that allows evaluation of features such as cell morphology and polarization, energy metabolism, signal transduction, and neurotransmitter release. 1
For many cell types, the introduction of nucleic acids and other substrates has become a groundbreaking tool to explore gene function. For example, RNA interference (RNAi), a form of posttranscriptional gene silencing, provides a useful tool for generating gene knockout model systems and studying pathways and gene and protein function in cell culture.3,4 The key to successful RNAi experiments is efficient delivery of substrates into cells. 5 Nondividing and differentiated cells, including primary neurons, are refractory to traditional nonviral methods such as lipid-mediated transfection. In addition, results are unsatisfactory with regard to transfection efficiency and/or cytotoxicity. Viral-mediated gene transfer, although highly efficient, is expensive, time-consuming, and requires safety precautions.6,7 The obvious lack of simple and efficient methods for transfection of primary neurons has been a bottleneck in neuroscience for years. Advances have been made in the field of electroporation, where a voltage pulse temporarily alters the properties of the plasma membrane, allowing extracellular material to enter the cell.8–11 High transfection efficiencies can be achieved with conventional electroporation but often at the expense of cell viability and survival.12–14
One electroporation-based approach for transfection of primary neurons is nucleofection. This is a modified form of cuvette electroporation, using a series of high-voltage pulses that enable plasmids to directly enter the nucleus.7,15 Optimized buffers and cell type–specific pulse protocols minimize damage to the cellular membranes, resulting in higher transfection efficiencies and cell survival than conventional electroporation. The main limitation of cuvette electroporation and transfection of neurons is that cells have to be transfected immediately after isolation, eliminating applications involving mature neurons with developed axons, dendrites, and functional synapses. Furthermore, nucleofection requires several liquid-handling steps that at present cannot be fully automated. The Cellaxess Elektra (Cellectricon AB, Mölndal, Sweden) technology uses a capillary electroporation concept that can be used for introduction of different types of molecules to adherent cells in 384-well plates. These range in size from target-designed dyes to oligonucleotides, small interfering RNA (siRNA), and plasmids to adherent cells in 384-well plates. A focused electrical field minimizes electrochemical toxicity and Joule heating, and differentiated cells such as primary neurons can be transfected in any developmental state with retained morphology and excellent viability. The Cellaxess Elektra is the only available electroporation method that enables fully automated transfection of primary neurons with high efficiency.
Materials and Methods
Primary Neuron Culture
Fresh embryonic day 18 rat brains (combined hippocampus and cortex) (Brainbits LLC, Springfield, IL) were triturated with small- and large-bore pipets in B27/Hibernate medium (Brainbits LLC). After trituration, undispersed pieces were allowed to settle, and the supernatant was transferred to new tubes and centrifuged at 200 g for 1 min. The pellet was resuspended in B27/Neurobasal with 0.5 mM Glutamax (Invitrogen, Carlsbad, CA). Cells were counted using the NucleoCounter (New Brunswick Scientific, Edison, NJ) and plated in Cellaxess Elektra 384-well plates coated with poly-D-lysine (Cellectricon AB). Cultures were maintained at 37 °C and 5% CO2 for 5 days.
Transfection
For transfection with a plasmid vector coding for CopGFP (Eurogen, Moscow, Russia), a source plate (AbGene, Surrey, UK) with plasmid diluted to 100 ng/µL in Cellaxess Elektra Accelerator Solution (Cellectricon AB) was prepared and loaded onto the instrument together with the cell plate, a storage plate for recycled medium, and a trough containing fresh cell culture medium. For siRNA experiments, siGFP (cat AM4635) or scrambled siRNA (cat AM2646) from Ambion (Life Technologies, Paisley, UK) at a final concentration of 100 nM was diluted together with pMOWS green fluorescent protein (GFP) plasmid. 16 The Cellaxess Elektra protocol for transfection was started through the Commander software. The automated transfection process was composed of the following work steps; 30 µL of the culture medium was transferred to the storage plate, leaving 10 µL of residual medium. Then, 40 µL Cellaxess Elektra Accelerator Solution with plasmid was added, and the electroporation protocol was carried out. Next, 15 µL Accelerator Solution was removed and 25 µL recycled culture medium was added back to the cells. Finally, 15 µL fresh medium was added to each well, and the cell plate was returned to the incubator. When optimizing Cellaxess Elektra transfection, 16 different pulse protocols were applied in a single 384-well plate (24 wells per condition), making the optimization procedure very efficient.
Immunocytochemistry
Twenty-four hours posttransfection, cells were fixed, permeabilized, and stained. Briefly, fixation and nuclear staining was performed for 15 min using 40 µL/well of 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) and 10 µg/mL Hoechst 33342 (Invitrogen, Eugene, OR) in phosphate-buffered saline (PBS), pH 7.4 (Invitrogen, Carlsbad, CA). Cells were rinsed two times with PBS followed by permeabilization for 20 min in 40 µL/well of 0.25% Triton-X100 (Sigma, St. Louis, MO) and 1% goat serum (Santa Cruz Biotechnology, Santa Cruz, CA). Permeabilization buffer was removed and cells were incubated for 1 h at 37 °C in 20 µL/well mouse anti-MAP-2 primary antibody (Santa Cruz Biotechnology or Abcam, Cambridge, UK) in permeabilization buffer. The plates were washed two times with PBS and 0.25% Triton-X and incubated for 1 h with 20 µL/well Texas Red X–conjugated goat antimouse secondary antibody (Santa Cruz Biotechnology or Abcam) diluted in permeabilization buffer. The fixed and stained cell plates were washed two times with PBS and 0.25% Triton-X100 and left in 50 µL/well PBS.
Imaging and Analysis
For plasmid transfection efficiency measurements, the plates were analyzed by an ImageXpress Micro (Molecular Devices, Sunnyvale, CA). We captured four images per well in three channels: (a) nuclei (Hoechst), (b) neuron cell body and neurites (MAP-2), and (c) GFP. Images were processed and analyzed using the MetaXpress software (Molecular Devices).
Figure 1b
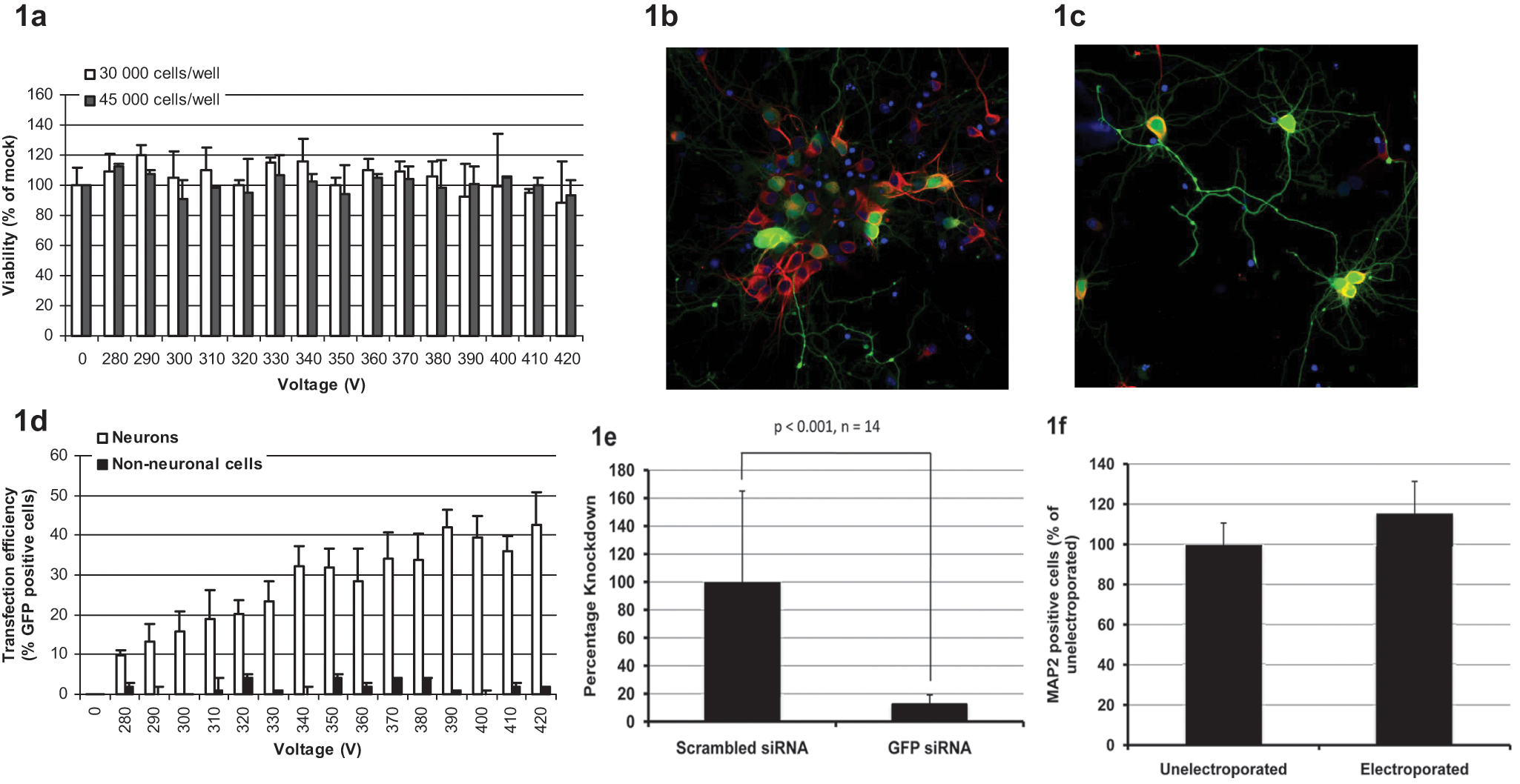
Optimization of transfection in primary rat neurons. (
Results and Discussion
Dissociated embryonic neurons were cultured in poly-D-lysine-coated 384-well Cellaxess Elektra plates at densities between 3000 and 45 000 cells (neurons and glia) per well. After 5 days in culture, neurons looked healthy and had a differentiated morphology with visible and extensive neurites. The fraction of neurons in culture was approximately 50% as measured by nuclear stain and MAP-2 (not shown). To make sure that the differentiated neurons could tolerate the liquid-handling steps associated with Cellaxess Elektra transfection, some plates were processed without any applied voltage. Processed plates could not be distinguished from unprocessed plates in terms of morphology of neurons or numbers of cells, as measured by immunostaining (MAP-2) and nuclear count.
In the neuron experiments, two pulses lasting for 2 ms each were delivered with a 15-s interval. Voltages ranging between 270 and 410 V were applied, and viability was measured 24 h following electroporation by means of counting the number of MAP-2-stained neurons compared with mock electroporation. In the absence of plasmid, neuron viability was >85% in all cases, showing that Cellaxess Elektra electroporation is very well tolerated by these cells, independent of cell density ( Fig. 1a ). The very low electrochemical toxicity and the fact that no trypsinization or replating steps are needed ensure that cell consumption is significantly reduced compared with other electroporation techniques,12,14,15 something that is important to take into account when screening a large number of genes in cell types that are challenging or expensive to prepare.
The same array of voltages was applied in the presence of plasmid, and GFP expression was studied 24 h after transfection. MAP-2 staining was used to distinguish neurons from glia. Transfected neurons expressed high levels of GFP in the cell body as well as in the extensions ( Fig. 1b , c ). Differentiated neurons are a valuable model for studying mechanisms involved in neuronal degeneration, axonal transport, or synaptic plasticity. The main drawback of other electroporation methods is that cells can only be transfected directly after isolation, eliminating the possibility of genetic manipulation of polarized neurons.6,7 In our experiments, GFP expression co-localized with MAP-2, indicating that transfection is neuron specific. Transfection efficiency was determined by dividing the number of GFP-positive neurons with the total number of neurons. When using 30 000 cells per well, the best protocols resulted in transfection efficiencies between 40% and 50% ( Fig. 1d ). At 3000 cells per well, the fraction of GFP-positive neurons was 20.2% ± 8.9%, showing that it is possible to further reduce the number of cells needed for large-scale experiments. The fraction of GFP-expressing nonneuronal cells was very close to zero ( Fig. 1d ), confirming that the transfection protocol is indeed specific for neurons. When optimizing parameters for in situ electroporation, several different parameters are important, including cell size, shape, thickness, and origin, making it possible to transfect a specific cell type in mixed cultures. The Cellaxess Elektra electroporation protocols are cell specific but not substrate specific, making it possible to co-transfect with both plasmids and siRNA or to change substrates without reoptimization steps. When adding siGFP together with the plasmid, GFP expression was significantly reduced 72 h after transfection compared with when using scrambled siRNA and plasmid at 350 V ( Fig. 1e ). The number of viable neurons 72 h after electroporation was unaffected compared with nonelectroporated controls ( Fig. 1f ), showing that differentiated neurons are viable for several days after transfection.
Biological variance is present in all research conducted in living systems. Further variance is expected when manipulating every gene in the genome, so the total variance on the readout of interest can be massive. This can be minimized by using biologically relevant models and complex readouts for RNAi screens. 17 The Cellaxess Elektra plates are fully compatible with most high-content analysis platforms, making it possible to do multifaceted high-content readouts. Primary cells are desired for gene silencing experiments because they better model the in vivo cells than immortalized cell lines. 14 In comparison to electroporating single samples, multiple transfections at a time significantly reduce handling time and variability, and with the Cellaxess Elektra transfection, efficiencies are robust between runs and across the 384-well plates ( Fig. 2 ).
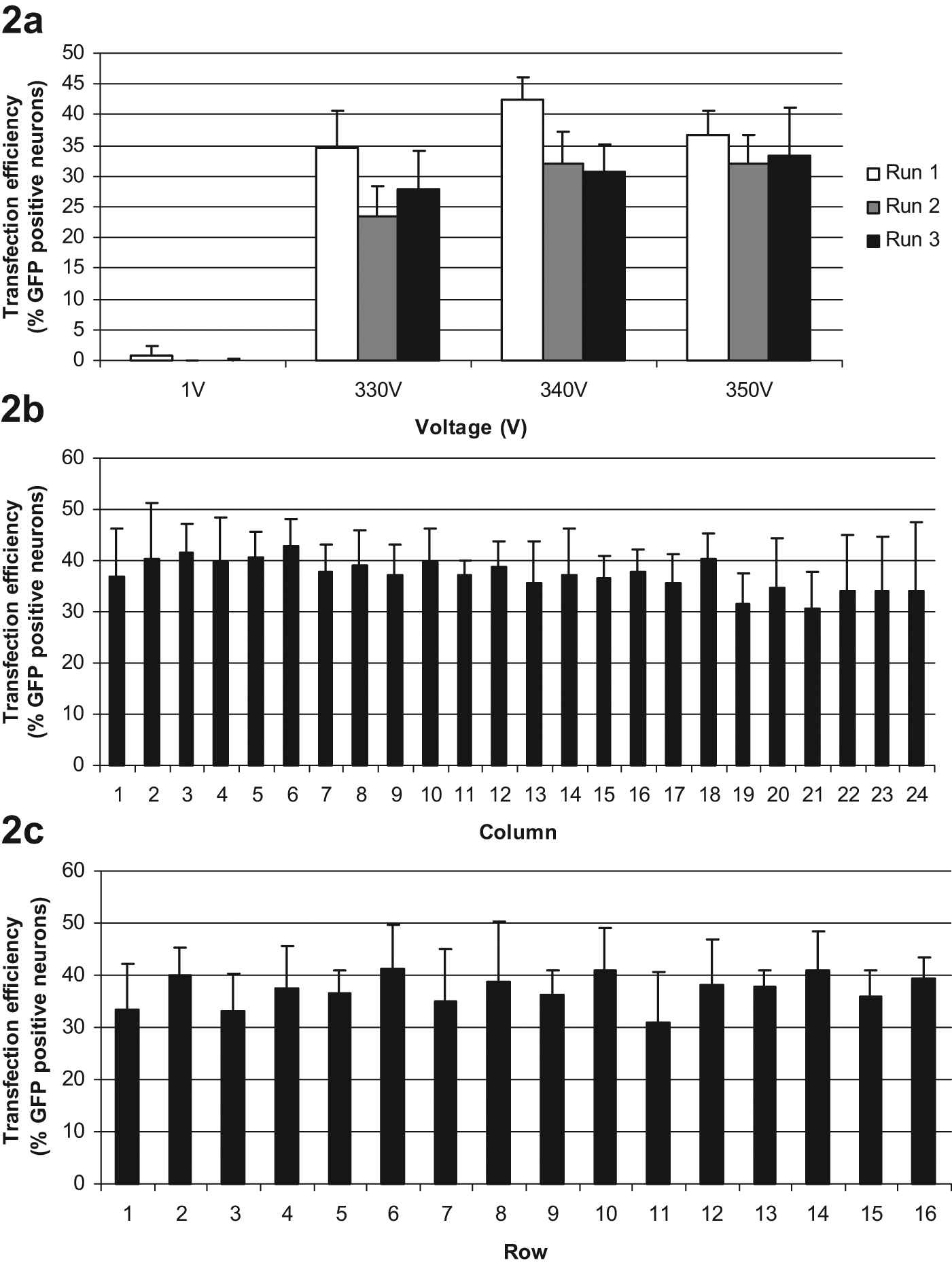
Transfection robustness. (
Previously described electroporation methods such as nucleofection have enabled genetic manipulation of neurons to some extent, but they are not appropriate for screening hundreds or thousands of genes since cell consumption and handling time are extensive. Furthermore, differentiated primary neurons with established neuronal networks have been difficult to target with these methods. The fact that Cellaxess Elektra delivers genetic material in situ to as many as 384 samples at a time enables high-throughput screening for hard-to-transfect primary cells.
Footnotes
Acknowledgements
We thank the Centre for Cellular Imaging at the University of Gothenburg for technical help and expert advice.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.