
Abstract
Von Hippel–Lindau (VHL) disease is an autosomal dominant disorder that affects multiple organs. Treatment is mainly surgical, and effective systemic therapies are needed. We developed a cell-based screening tool to identify compounds that stabilize or upregulate full-length, point-mutated VHL protein. The 786-0 cell line was infected with full-length W117A-mutated VHL linked to a C-terminal Venus fluorescent protein. This VHL-W117A-Venus line was used to screen the Prestwick drug library and was tested against proteasome inhibitors MG132 and bortezomib. Western blot validation and evaluation of functional readouts, including hypoxia-inducible factor 2α (HIF2α) and glucose transporter 1 (Glut1) levels, were performed. We found that bortezomib, MG132, and the Prestwick compounds 8-azaguanine, thiostrepton, and thioguanosine upregulated VHL-W117A-Venus in 786-0 cells. 8-Azaguanine downregulated HIF2α levels and was augmented by the presence of VHL W117A. VHL p30 band intensities varied as a function of compound used, suggesting alternate posttranslational processing. Nuclear-cytoplasmic localization of VHL-W117A-Venus varied among the different compounds. In conclusion, a 786-0 cell line containing VHL-W117A-Venus was successfully used to identify compounds that upregulate VHL levels, with differential effect on VHL intracellular localization and posttranslational processing. Further screening efforts will broaden the number of pharmacophores available to develop therapeutic agents that will upregulate and refunctionalize mutated VHL.
Introduction
Identification of von Hippel–Lindau syndrome occurred through the observation by Eugen von Hippel, an ophthalmologist, of familial retinal hemangioblastomas in 1904 and Arvid Lindau, a neurologist, of hereditary hemangioblastomas in 1927. 1 As more individuals with this syndrome were recognized, involvement of the kidney, with renal cysts and clear cell renal cell carcinoma (RCC), was reported, as was involvement of the adrenal gland with pheochromocytomas and the pancreas with cysts, serous cystadenomas, and pancreatic neuroendocrine tumors. In 1993, Latif and colleagues published the identification of the VHL gene, 2 and in the intervening years, a large amount of research has been performed to elucidate the function of the VHL protein (VHL). Recognition that VHL is an E3 ubiquitin ligase that forms a complex with elongins C and B, 3 as well as Cullin 2, 4 enabling the mature VHL–elongin C and elongin B (VBC) complex to bind and postranslationally regulate hypoxia-inducible factors 1α and 2α (HIF1α and HIF2α), 5 and that this process required prolyl hydroxylation of HIF1α and HIF2α6 provided a better understanding of the highly vascular nature of most lesions arising in the background of a VHL mutation. The discovery of VHL’s HIF regulatory capacity and the impact of unbridled HIF expression on the expression of multiple proangiogenic factors, including vascular endothelial growth factor (VEGF), led to the development of multiple therapeutic agents targeting the VEGF pathway in neoplasms.7–10 Although these agents have demonstrated biological and clinical activity in neoplastic diseases, including clear cell RCC, which in both the germline and sporadic setting demonstrate near-universal inactivation of VHL, there have been few recorded examples of complete tumor regression or cure. Even more troubling has been the near-complete lack of activity of these agents in hemangioblastomas, a highly vascular but noninvasive or nonmetastasizing lesion that arises in the eye, cerebellum, brainstem, and spinal cord of patients with VHL syndrome.
Reasons for the lack of greater benefit from VEGF axis–blocking agents have not been fully elucidated. Possibilities include redundancy in the proangiogenic signaling network, with both innate and acquired resistance pathways likely present. The elucidation of a number of novel functions for VHL, including regulation of the primary cilium 11 and the cilia centrosome cycle, as well as regulation of extracellular matrix deposition via binding to collagen IV 12 and fibronectin, 13 demonstrates that management of the HIF-related effects of VHL loss may not be sufficient to reverse the neoplastic phenotype in germline or sporadic cases of VHL-related diseases.
In an effort to better understand the pathogenesis of VHL mutations, genotype-phenotype correlations have been made between specific classes of VHL mutations and their clinical and biological significance. VHL is a 213–amino acid protein with three exons14,15 and an internal start site at amino acid 54, resulting in the expression of a 19-kDa VHL p19 isoform and the full-length VHL p30 isoform. 16 As of now, there have been virtually no recognized nonpathogenic polymorphisms. 17 Approximately one-third of all VHL mutations are missense, resulting in the generation of a full-length protein. 15 Data demonstrate that point-mutated VHL is less stable than wild-type protein 18 and is rapidly cleared from the cell via heat shock protein–mediated proteasomal degradation,19,20 despite maintaining residual functionality in some cases. 18 In addition, there is a distinct subset of VHL mutations that render nascent VHL incapable of binding to the eukaryotic type II chaperonin tail-less complex polypetptide–1 (TCP-1) ring complex (TRiC), also called CCT for chaperonin-containing TCP-1. 21 These mutations occur in amino acids 114 to 119 and 148 to 155 and constitute the two major TRiC-binding domains, named Box 1 and Box 2, respectively. 19 By failing to bind to TRiC, VHL cannot fold into its mature form and cannot generate a mature VBC complex. Furthermore, disease-causing mutations in the region spanning amino acids 155 to 181 demonstrate decreased binding of elongin C. 3 Failure to load elongins C and B onto VHL results in the failure of VHL release from TRiC and more rapid degradation of VHL. 22
We hypothesized that compounds that restabilize mutant VHL, either through decreased proteasomal degradation or via the facilitation of refolding and proper VBC complex formation, would aid in the reacquisition of a normalized cellular phenotype. To discover compound leads that could test our hypothesis, we generated VHL-deficient 786-0 cell lines that contain Venus 23 high-intensity fluorescence-tagged VHL with a highly unstable W117A mutation (VHL-W117A-Venus), evaluated VHL-W117A-Venus levels after proteasome inhibitor treatment, and screened the 1200 compound Prestwick library for other compounds that manipulate VHL-W117A-Venus levels. We discovered three compounds that were able to reliably upregulate VHL-W117A-Venus levels. Our data also provide an intriguing new mechanism of action for bortezomib, a proteasome inhibitor in clinical use that has shown some efficacy in RCC, and the ability of the VHL-W117A-Venus-containing cell line to be used as an assay to successfully identify candidate molecules that consistently increase intracellular VHL levels.
Materials and Methods
Cell culture and plasmids
RCC 786-0 and HEK293T cell lines were from ATCC (Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle’s medium from Invitrogen (Carlsbad, CA), supplemented with 10% fetal calf serum from Gibco (Carlsbad, CA). VHL-wt-GFP plasmid was from Dr. Paul Corn (MD Anderson Cancer Center, Houston, TX). Venus-expressing plasmid Venus/pCS2 was from Dr. Atsushi Miyawaki (RIKEN Brain Science Institute, Saitama, Japan). VHL-wt-Venus was made by replacing green fluorescent protein (GFP) in VHL-wt-GFP with Venus from Venus/pCS2 as a BamHI/XbaI fragment. VHL-W117A-Venus mutation fusion was made from VHL-wt-Venus by mutagenesis with the QuikChange mutagenesis kit from Stratagene (La Jolla, CA) and confirmed by sequencing. (Primers for the mutagenesis: GCTACCGAGGTCACCTTGCGCTCTTCAGAGATGCAGG and CCTGCATCTCTGAAGAGCGCAAGGTGACCTCGGTAGC.) Retroviral vector pLEGFP-N1 was from Clontech (Mountain View, CA). Retroviral expression plasmids VHL-wt-Venus-Retro and VHL-W117A-Venus-Retro were made by moving the VHL-wt-Venus or VHL-W117A-Venus gene fragment into pLEGFP-N1 to replace GFP. VHL-W117A was made by digestion of VHL-W117A-Venus with BamHI/XbaI followed by blunting and religation to remove Venus. Cells were transfected using FuGENE 6 transfection reagent from Roche (Indianapolis, IN) following the manufacturer’s protocol. Retrovirus preparation and infection were performed as previously described. 24
Reagents and antibodies
MG132 was from Sigma (St. Louis, MO). Bortezomib was from Selleck Chemicals (Houston, TX). Candidate drugs were from Prestwick (Prestwick Chemical, Illkirch, France). Restriction enzymes and T4 DNA ligase were from New England Biolabs (Ipswich, MA). Western blotting antibodies for VHL (#2738) and IGF1R (#3018) were from Cell Signaling Technology, Inc. (Beverly, MA). Antibody for HIF2α (#NB100-122) was from Novus Biologicals (Littleton, CO). Antibody for Glut1 (#GT12-A) was from Alpha Diagnostic International (San Antonio, TX). GAPDH (#AM4300) was from Ambion (Austin, TX).
Western blotting and cell proliferation assay
To prepare cell lysates for Western blotting, cells were lysed in RIPA buffer (Tris-HCl [pH 7.4], 50 mM; NaCl, 150 mM; NP-40, 1%; sodium deoxycholate, 0.5%; sodium dodecyl sulfate [SDS], 0.1%) supplemented with protease inhibitor and phosphatase inhibitor cocktail (Pierce, Rockford, IL). Western blotting was performed as previously described. 24 Cell proliferation was determined by the CellTiter Blue Cell Viability assay from Promega (Madison, WI) according to the manufacturer’s protocol.
High-throughput microscopy and image analysis
For the Prestwick compound screen, VHL-W117A-Venus 786-0 cells were plated at 6000 cells in 100 µL per well of complete culture media in black Corning (Corning Life Sciences, Lowell, MA) 96-well optical plastic bottom plates and incubated an additional 12 to 18 h to allow for cell adhesion and spreading. Compound dilutions and final addition to multiwell plates were performed using the multichannel pod of a Beckman Biomek FX robotic platform (Beckman Coulter, Brea, CA) to ensure repeatability from experiment to experiment. Compounds from the Prestwick Chemical Compound Library (Prestwick Chemical) were used for analysis. The Prestwick library comprises 1200 off-patent compounds that have been approved by the Food and Drug Administration (FDA) in the past for various indications. These compounds were initially diluted 10-fold from a stock containing 2 mg/mL in DMSO with cation-free phosphate-buffered saline (PBS; MediaTech, Inc., Manassas, VA). The test compounds were further diluted 20-fold by addition to assay wells. Basal expression levels of VHL-W117A-Venus were determined in cells treated with PBS containing a final concentration of 0.5% DMSO. The agonist-induced phenotype was determined in control wells treated with 0.5 µg/mL N-(benzyloxycarbonyl) leucinyl-leucinyl-leucinal (Z-Leu-Leu-Leu-al or MG132) in 0.5% DMSO. After incubation for an additional 20 to 23 h, plates were washed with PBS and fixed for 20 min at room temperature in 4% paraformaldehyde in PBS (Affymetrix-USB, Santa Clara, CA). After fixation, cells were briefly permeabilized (5 min) with 0.5% Tween 20 (v/v) and prepared for imaging by washing in PBS, removing the wash solution, and adding a 1-µg/ml DAPI solution, staining DNA to facilitate nuclear image segmentation. Cells were imaged in PBS.
Cells were imaged using the Beckman Cell Lab IC-100 Image Cytometer (Beckman Coulter) platform that consists of (1) a Nikon Eclipse TE2000-U inverted fluorescence microscope (Nikon, Melville, NY), (2) a Chroma 82000 triple band filter set (Chroma, Brattleboro, VT), (3) a Hamamatsu ORCA-ER Digital CCD imaging camera (Hamamatsu, Bridgewater, NJ), and (4) a Photonics COHU Progressive scan focusing camera (Photonics, Oxford, MA). The microscope was equipped with a Nikon S Fluor 20×/0.75NA objective and the imaging camera set to capture 8-bit images at 2 × 2 binning (1344 × 1024 pixels; 0.42 µm2 pixel size) with two channels captured per field: channel 0 (DAPI; ex 350/50 nm, em 420 nm/long pass) was used to find the focus and cell nuclei, and channel 1 was used to image VHL-W117A-Venus (Venus; ex 490/20 nm, em 517/30 nm). In general, 16 fields were captured per well for image analysis.
Images were analyzed using Cytoshop Version 2.1 (Beckman Coulter, Indianapolis, IN) as described by Szafran et al. 25 Image processing began by applying a general automatic mean background subtraction in each of the channels and the removal of partial cells visible at the edges of each frame. Next, cells were identified by using nuclear masks generated by a combination of image filters and automatic histogram-based thresholding. Total area of the cells was determined by intersection of a chosen extraction radius (approximately 25% larger than average nucleus radii) and tessellation polygons generated by the software. Agonist-induced VHL-W117A-Venus fluorescence levels were determined as the total integrated pixel intensity in the GFP channel (CORR1). All values were corrected for background VHL-W117A-Venus fluorescence measured in the DMSO-only control wells. Candidate compounds were chosen using the following criteria: (1) fluorescence level at least 50% of corresponding MG132 control samples and (2) overall cell number at least 10% of DMSO control well number. The latter evaluation was designed to eliminate highly toxic compounds and readouts that are due to an excessively small sample size.
Results
Identification and Characterization of Unstable VHL-W117A-Venus Mutation
To generate a screening tool that permits identification of compounds that restabilize mutant VHL, we mutated codon TGG to GCG, which results in a tryptophan to alanine alteration at amino acid 117 within the Box 1 TRiC binding region. This sequence was placed in a pLEGFP-N1 retroviral vector, fusing VHL W117A to a C-prime Venus high-intensity fluorescent tag (VHL-W117A-Venus), and infected into the VHL–/– 786-0 cell line. A similar strategy was used to generate a 786-0 line containing Venus-tagged wild-type VHL (VHL-wt-Venus). Comparison of fluorescence intensity levels between VHL-W117A-Venus and VHL-wt-Venus demonstrated a significant decrease in mean fluorescence intensity of the VHL-W117A-Venus cells despite similar cell confluence ( Fig. 1A ). Protein level of VHL-W117A-Venus was significantly lower than that of VHL-wt-Venus examined by Western blotting ( Fig. 1B ). Levels of HIF2α and glucose transporter 1 (Glut1), a HIF-regulated gene product, were decreased by wild-type but not mutant VHL ( Fig. 1B ). Growth rates of 786-0 expressing VHL-W117A-Venus were similar to parental cells ( Fig. 1C ).
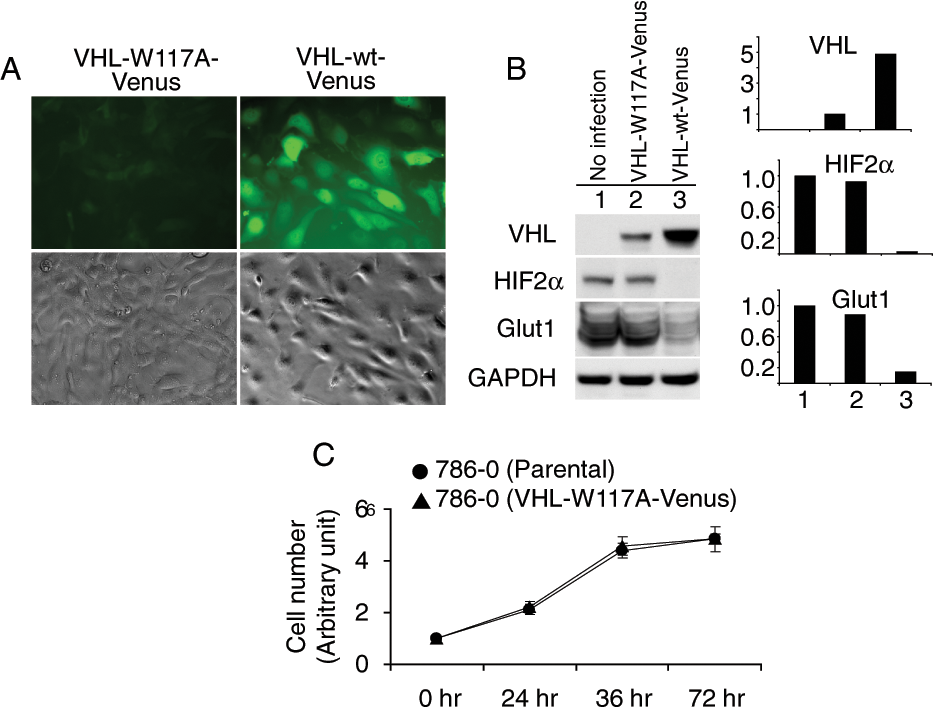
Von Hippel–Lindau (VHL) stability affected by W117A mutation. (
We then tested whether a proteasome inhibitor could upregulate VHL levels in VHL-W117A-Venus-containing 786-0 cell lines. As seen in Figure 2A , the addition of MG132 or bortezomib resulted in a significant increase of fluorescence levels. To confirm that VHL protein levels were increased, we performed Western blots on MG132- and bortezomib-treated samples and demonstrated an increase in VHL-W117A-Venus levels ( Fig. 2B ). Both upper and lower bands of VHL protein (arrow designates the upper band, and arrowhead designates the lower band) were upregulated, with a predominant increase in the lower band. Increased wild-type VHL levels were also noted after proteasome inhibitor treatment. HIF2α is one of the primary downstream substrates of VHL. Increased HIF2α levels were detected in MG132- and bortezomib-treated VHL-W117A-Venus cells ( Fig. 2B ), whereas Glut1 levels did not change. Wild-type VHL fully suppressed HIF2α and nearly completely suppressed Glut1. Ubiquitinated proteins were significantly increased in MG132- or bortezomib-treated samples, indicating proteasomal degradation was effectively inhibited ( Fig. 2B ).
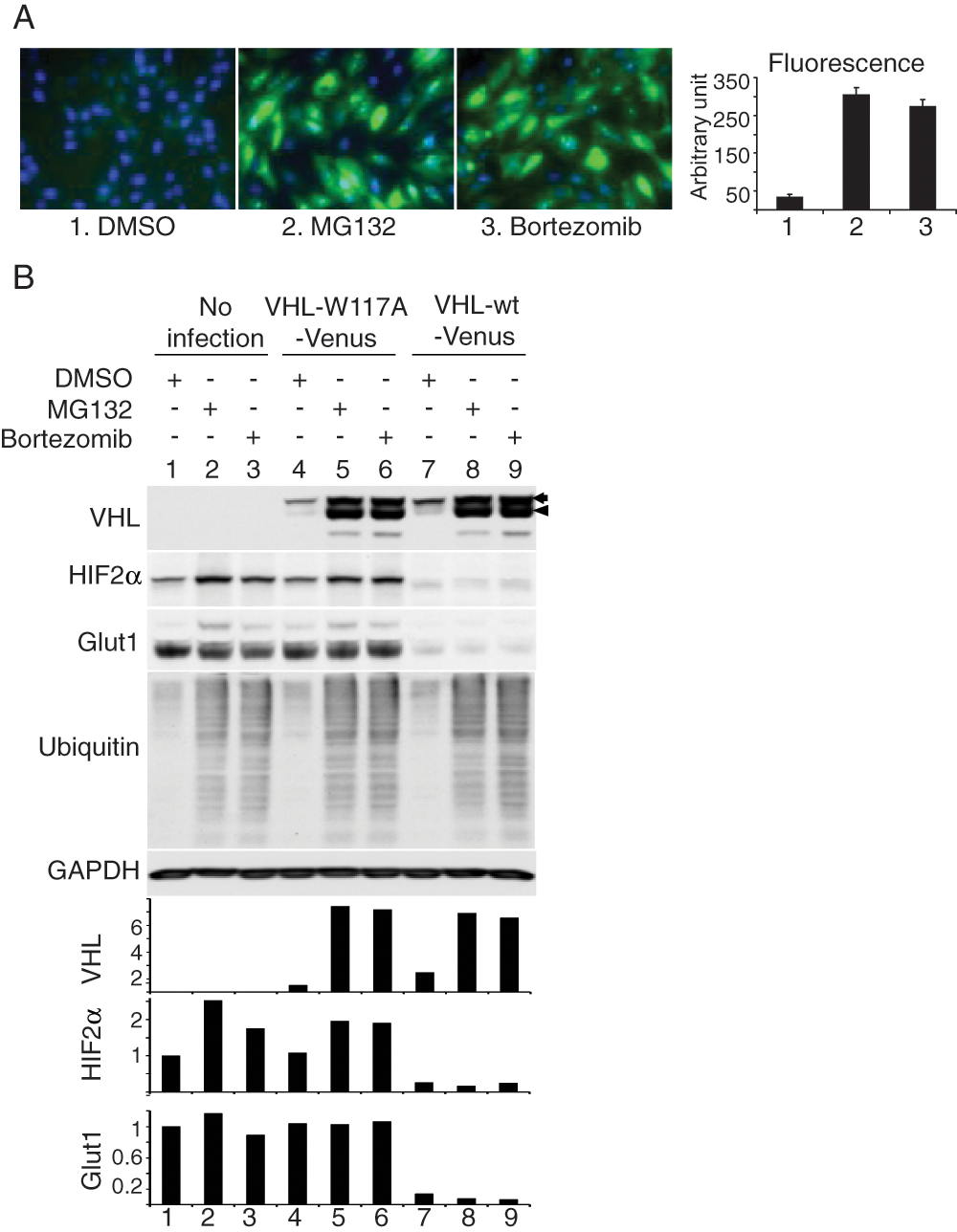
Effect of proteasome inhibition on stability of VHL-W117A-Venus. (
High-Throughput Screen to Detect VHL Modulating Compounds
To screen for compounds that upregulate mutated VHL levels, we generated a 96-well-based system using 786-0 cells expressing VHL-W117A-Venus and tested the Prestwick 1200-compound library. Figure 3A shows a representative plate result. MG132-stimulated fluorescence levels of VHL-W117A-Venus protein were on average 61-fold greater than background levels (signal-to-background ratio). Overall assay quality was determined using the Z′ calculation. A mean Z′ factor of 0.5 (n = 34) was determined from all primary screening plates.
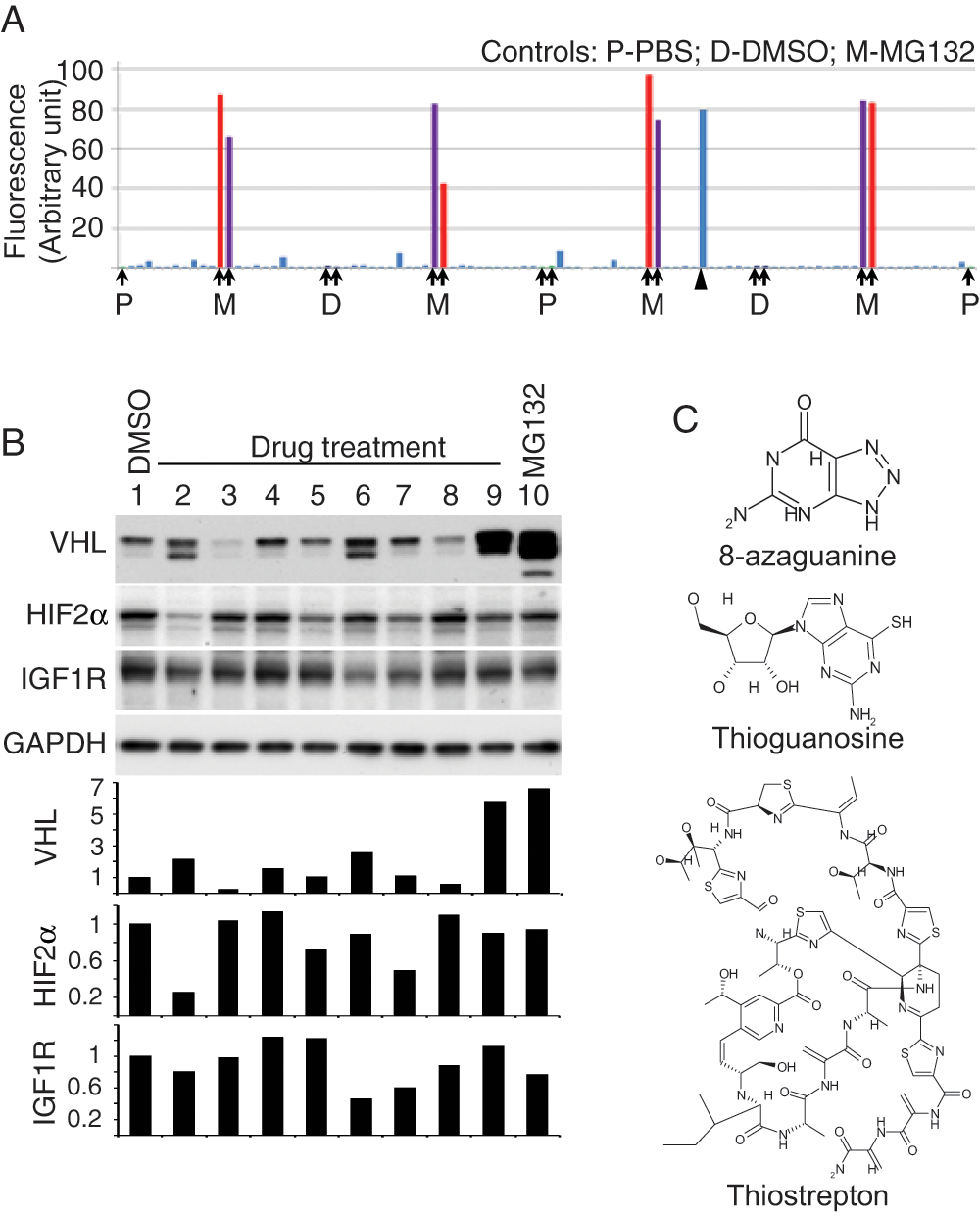
Proof-of-concept screen and candidate validation. (
Candidates were chosen on the basis of cellular fluorescence intensity as well as on relative viability of cells after treatment with compound (data not shown), resulting in the identification of eight “hit” compounds that increased fluorescence. These compounds included 8-azaguanine, R(–) apomorphine hydrochloride hemihydrate, dyclonine hydrochloride, ethacrynic acid, thioguanosine, colchicine, lovastatin, and thiostrepton. Candidate compounds were validated by evaluating levels of VHL protein after treatment. As seen in Figure 3B , compounds in lanes 2, 6, and 9 demonstrated substantial upregulation of VHL levels. Compound in lane 2 is 8-azaguanine, compound in lane 6 is thioguanosine, and compound in lane 9 is thiostrepton ( Fig. 3C ). These compounds predominantly upregulated the lower of the two VHL bands.
Functional Assessment of Candidate Compounds
To better understand the impact of compound-induced upregulation on VHL function, we assessed the impact of 8-azaguanine, thioguanosine, and thiostrepton on VHL effector protein levels. As shown in Figure 3B , HIF2α was significantly downregulated by 8-azaguanine in 786-0 VHL-W117A-Venus cells. Compared with DMSO controls, thioguanosine, thiostrepton, and MG132 did not affect HIF2α levels significantly. Insulin-like growth factor 1 receptor (IGF1R) has also been shown to be regulated in a VHL-dependent fashion, with higher levels of functional VHL associated with decreased IGF1R levels. 26 As seen in Figure 3B , consistent downregulation of IGF1R was not seen with increasing VHL levels. To assess effect of candidate compounds on untagged VHL W117A, HEK293T cells were transiently transfected to express VHL-W117A without tag. The same proportional drug effect was seen ( Fig. 4A ). To better understand the kinetics of the VHL increase as a function of treatment time, the time course of VHL increase was assessed in 786-0 VHL-W117A-Venus cells ( Fig. 4B ). Total levels increased over the 24-h period, with differential upregulation of the upper versus lower VHL band. Further evaluation of the significance of the two bands revealed that phosphatase treatment (data not shown) did not affect either band, suggesting that posttranslational modifications other than phosphorylation are responsible for the different bands. In addition, transfection of 786-0 cells with the p19 form of VHL lacking the 53 N-terminal amino acids confirms a 19-kDa size and rules out the possibility that this additional band is due to known internal translation start site–dependent regulation (data not shown). Further evaluation of the 8-azaguanine effect on HIF2α levels in the parental 786-0 line revealed that HIF2α levels also decreased in a dose-dependent and VHL-independent fashion ( Fig. 4C ), suggesting that a VHL-independent mechanism of action for the effect of 8-azaguanine on HIF2α level is also operative. To better understand the impact of the compounds on VHL intracellular localization, we evaluated fluorescent images of VHL-W117A-Venus 786-0 cells (Fig. 4D). These reveal different VHL distribution patterns: 8-Azaguanine results in a fairly homogeneous cytoplasmic and nuclear distribution. Thioguanosine treatment results in an intriguing nuclear exclusion of VHL, and thiostrepton induces a predominant nuclear distribution.
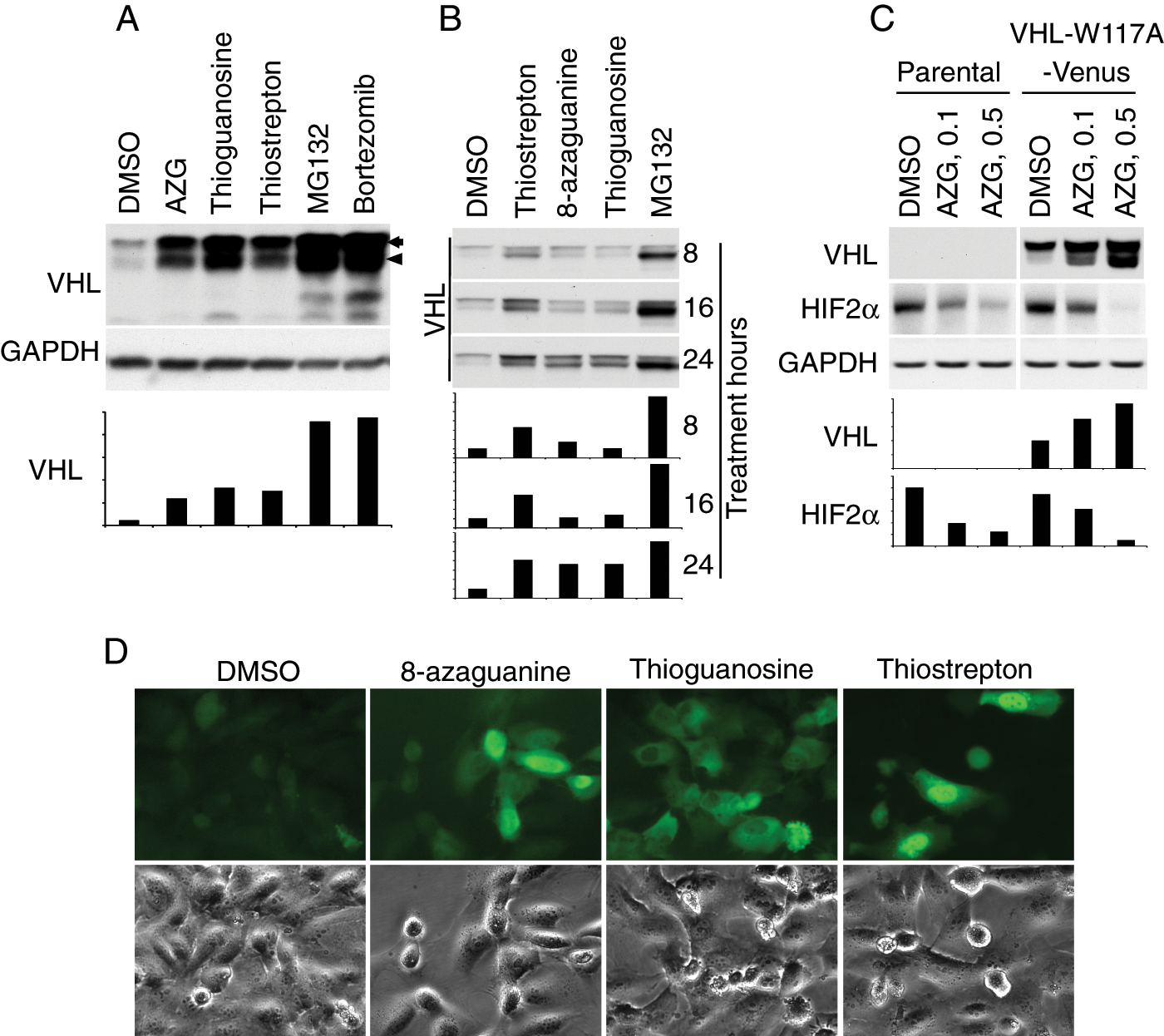
Functional assessment of candidate compounds. (
Discussion
Von Hippel–Lindau disease is a devastating illness with no satisfying therapeutic options. A subset of VHL mutations possesses residual functionality, but these mutations are unstable due to their recognition by the proteostatic regulatory machinery and are rapidly degraded.
An overarching goal of our effort was to develop a set of reagents that are capable of influencing VHL proteostasis. Nascent VHL is shuttled to the TRiC/CCT chaperonin via heat shock protein 70, where it is folded into a mature form and where elongin C and B loading occurs. 19 Once the VBC complex is formed, it is released from TRiC and performs its function as an E3 ubiquitin ligase, as well as other key activities. Mutations in amino acids 114 to 119 and 148 to 155 interfere with TRiC binding, resulting in a failure of proper folding and decreased VBC complex formation. 19 Mutations in amino acids 155 to 181 decrease elongin C binding 3 and decrease VHL’s ability to regulate HIF isoforms. Compounds that can improve the interaction between mutated VHL and TRiC, or the loading of the elongin C and B proteins onto TRiC-bound VHL, may improve the phenotype of point-mutated VHL cells and alter disease behavior.
We developed a fluorescence-based high-throughput screening tool to detect compounds that upregulate intracellular levels of mutated VHL-W117A-Venus and performed experiments to explore the functional consequences of VHL-W117A-Venus upregulation. We first hypothesized that a proteasome inhibitor would provide an effective positive control for a VHL stability-inducing agent. Indeed, both the laboratory-grade MG132 and the FDA-approved bortezomib were capable of upregulating VHL-W117A-Venus levels. It is important to note that two VHL bands are seen. The significance of the double bands is not known, but it is interesting to note that the lower band is preferentially upregulated by proteasome inhibitors and some of the screen compounds. Further analysis of the differentiating characteristics between these two bands may shed light on the significance of this finding. After MG132 and bortezomib treatment, we observed a slight increase in HIF2α levels, which is expected in the face of effective proteasomal blockade and is confirmed with ubiquitin blots. However, these data also suggest a lack of functional normalization of VHL-W117A-Venus, despite increases in VHL levels, since VHL-wt-Venus-containing cells were capable of maintaining suppressed HIF2α and Glut1 levels in the face of proteasome inhibition ( Fig. 2B ).
Our high-throughput screen identified three compounds that consistently and robustly upregulated VHL-W117A-Venus from an initial set of eight candidates. As seen in Figure 3B , VHL-W117A-Venus levels were either the same or lower than control cells in five of the eight hits. This may be due to induction of other cellular processes that induce autofluorescence or to frank false positives. The true positives include the purine-derived antimetabolite chemotherapy agents 8-azaguanine and thioguanosine, as well as the thiazole antibiotic thiostrepton. A previously published screen identified 8-azaguanine as a lead compound capable of modulating HIF levels in neoplastic cells. 27 Thiostrepton has recently been recognized to act as a proteasome inhibitor. 28 This is consistent with the findings we observed with MG132 and bortezomib in our initial validation studies. In addition, the predominant nuclear localization of the VHL-W117A-Venus signal is similar in thiostrepton and MG132. It is less clear how 8-azaguanine and thioguanosine affect VHL levels. Figure 3B shows that with the exception of 8-azaguanine, higher cellular levels of VHL-W117A-Venus achieved with these compounds are insufficient to achieve consistent downregulation of HIF2α or IGF1R, suggesting that the compounds found in our screen do not consistently refunctionalize the mutant isoform. Nevertheless, effects on subcellular localization are observed. Figure 4A shows that the cellular distribution of VHL-W117A-Venus following 8-azaguanine and thioguanosine treatment is similar, with predominant cytoplasmic staining, although in the case of thioguanosine, it appears that there is nuclear exclusion of VHL-W117A-Venus. VHL performs a number of other non-HIF-dependent intracellular tasks, including p53 stabilization, 29 regulation of the primary cilium, 11 and alteration of collagen IV homeostasis. 12 Further analysis with readouts for these assays may further elucidate the impact these compounds have on VHL, and a larger screen will likely be required to identify compounds that are capable of both increasing VHL levels and having an effect on function.
In conclusion, generation of the VHL-W117A-Venus containing 786-0 line is a first step in the search for compounds that perform specific actions on VHL proteostatic machinery. Larger screens and more refined VHL-substrate interaction assays are necessary to achieve our broader goal of developing a panel of VHL proteostasis-modulating compounds.
Footnotes
Acknowledgements
The authors thank Deborah Ogutu for her editorial assistance.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This research is supported by a grant from NIH through the Common Fund (PN1EY016525).