
Abstract
In the present study, we developed a cell-based protocol for the identification of drugs able to induce steatosis. The assay measures multiple markers of toxicity in a 96-well plate format using high-content screening (HCS) technology. After treating HepG2 cells with increasing concentrations of the tested compounds, toxicity parameters were analyzed using fluorescent probes: BODIPY493/503 (lipid content), 2′,7′-dihydrodichlorofluorescein diacetate (reactive oxygen species [ROS] generation), tetramethyl rhodamine methyl ester (mitochondrial membrane potential), propidium iodide (cell viability), and Hoechst 33342 (nuclei staining). A total of 16 drugs previously reported to induce liver steatosis through different mechanisms (positive controls) and six nonsteatotic compounds (negative controls) were included in the study. All the steatosis-positive compounds significantly increased BODIPY493/503 fluorescence in HepG2 cells, whereas none of the negative controls induced lipid accumulation. In addition to effects on fat levels, increased ROS generation was produced by certain compounds, which could be indicative of increased risk of liver damage. Our results suggest that this in vitro approach is a simple, rapid, and sensitive screening tool for steatosis-inducing drugs. This conclusion should be confirmed by testing a larger number of steatosis-positive and -negative inducers.
Introduction
Steatosis, or increased lipid accumulation in the liver, mainly as triglycerides, can be induced by several factors, including exposure to certain drugs. 1 Excessive lipid accumulation is often reversible, and prolonged exposure to certain drugs can cause macrovacuolar steatosis, a benign hepatic lesion. However, in some patients, chronic macrovacuolar steatosis can evolve into steatohepatitis, and ultimately into cirrhosis. 2 Moreover, in a few patients, some drugs acutely induce microvesicular steatosis, which can potentially lead to liver failure with fatal consequences.1,2 These adverse effects have led to the interruption of clinical trials (i.e., fialuridine), postmarket withdrawal (i.e., perhexiline), or restricted use guidelines (i.e., valproic acid) of several drugs. 1 As the development of new drugs is a complex, lengthy, and expensive process that aims to identify pharmacologically active, low-toxicity drug candidates among closely related compounds, it could be advantageous to determine in early developmental stages which drugs are able to induce steatosis. To this end, simple, rapid, and predictive screening assays, in which many drug candidates are evaluated, are preferable approaches to identify and discard compounds with a strong steatosis-inducing potential.
In vitro assays to evaluate the steatogenic potential of new drugs are very scarce (revised by Amacher). 3 Nile Red or Bodipy dyes have been proposed as fluorescent probes to examine the accumulation of neutral lipids in liver-derived cultured cells.4,5 These fluorimetric assays are simple approaches to screen steatosis-inducing compounds. However, obtaining information on the potential mechanisms involved in hepatotoxicity due to steatogenic drugs requires the analysis of multiple endpoints. We recently reported a multiparametric flow cytometry assay to assess drug-induced steatosis in HepG2 cells in which the mechanistic information on hepatotoxicity caused by steatogenic compounds was obtained by using appropriate combinations of fluorescent probes. 6 The assay correctly identified 100% of steatosis-positive and steatosis-negative compounds and was suggested as a useful approach to identify new drugs with a clinically relevant steatotic potential. However, one major limitation of the procedure is that after treatment with the compounds, cells must be detached from culture plates prior to fluorescent staining and the flow cytometry analysis, which is time-consuming and notably limits the application of assays for high-throughput screenings of large sets of compounds.
In the present study, we propose a new cell-based protocol that aims to identify steatosis inducers. The assay is based on the analysis of a combination of toxicity endpoints (cell viability, intracellular lipid accumulation, mitochondrial membrane potential, hydrogen peroxide generation) using high-content screening (HCS) technology. By combining the use of sensitive, scarcely invasive, fluorescence-based multiparametric methods and the event-integrating concept of individual cells, HCS has been demonstrated to be a promising and valuable tool for the mechanistic understanding of drug-induced hepatotoxicity.7,8 Major advantages of this multiparametric test include high sensitivity, increased throughput, and use of adherent human cells cultured in 96-well formats.
Materials and Methods
Chemicals
The test compounds were purchased from the following sources: acetylsalicylic acid, amikacin hydrate, amiodarone hydrochloride, clofibrate, colchicine, cumene hydroperoxide, cyclosporine A, didanosine, doxycycline hydrochloride, DL-ethionine, fenofibrate, rotenone, stavudine, tamoxifen, tert-butyl hydroperoxide, tianeptine sodium salt, ticlopidine, tetracycline hydrochloride, and sodium valproate were acquired from Sigma (Madrid, Spain); sodium citrate from Merck (Barcelona, Spain); and fialuridine from Campro Scientific GmbH (Berlin, Germany); zidovudine from GlaxoSmithKline (Madrid, Spain). Fluorescent dyes BODIPY493/503, tetramethyl rhodamine methyl ester (TMRM), and 2′,7′-dihydrodichlorofluorescein diacetate (DHCF-DA) were purchased from Molecular Probes, Invitrogen (Madrid, Spain). Propidium iodide (PI) and Hoechst 33342 were purchased from Sigma.
Culture of HepG2 Cells
HepG2 cells (ECACC No.85011430) were cultured in Ham’s F-12/Leibovitz L-15 medium (1:1 v/v; Gibco BRL, Paisley, UK) supplemented with 5% newborn calf serum (Gibco BRL), 50 U penicillin/mL, and 50 µg streptomycin/mL. For subculturing purposes, cells were detached with 0.25% trypsin/0.02% EDTA (Gibco BRL) at 37 °C. For the steatosis-inducing studies, HepG2 cells were seeded into 96-well culture plates at a density of 5000 cells/well. External rows and columns were left without cells but contained culture media to protect the culture. Before treatments, cells were allowed to grow and equilibrate for 24 h.
Treatment of HepG2 Cells with Tested Compounds
Acetylsalicylic acid, amiodarone, clofibrate, cyclosporine A, didanosine, doxycycline, DL-ethionine, fenofibrate, fialuridine, stavudine, tamoxifen, tetracycline, tianeptine, ticlopidine, valproate, and zidovudine were selected as steatosis-positive inducers.2–6,9 In addition, amikacin, rotenone, t-butyl hydroperoxide, cumene hydroperoxide, colchicine, and citrate were included in the study as steatosis-negative compounds.6,10
Prior to being exposed to the tested compounds, HepG2 cells were preincubated for 14 h with a 62 µM mixture of oleate and palmitate (2:1 ratio), followed by a change to fatty-acid–free medium containing different concentrations of the test compounds for another 24-h period. 6 The stock solutions of compounds were prepared in DMSO or water and were diluted in the culture medium to obtain the desired final concentrations. The final DMSO concentration in the culture medium never exceeded 0.5% (v/v), and control cultures were treated with the same amount of solvent.
Multiparametric Assay Using HCS
After treatments, cells were washed and incubated with selected probes to measure the multiple parameters indicative of cell toxicity: intracellular fat deposits were detected by measuring BODIPY 493/503 fluorescence; accumulation of fluorescent compound 2′,7′-dichlorofluorescein (DCF), generated by intracellular oxidation of DHCF-DA, was used as it was indicative of reactive oxygen species (ROS) generation, primarily H2O2; the mitochondrial membrane potential was determined using TMRM fluorescence; cell viability was determined by PI exclusion; and Hoechst 33342 was used for nuclei staining and cell counts. Fluorescent dyes were combined according to their optical compatibility in a standard flow cytometer. 6 For each cell treatment (i.e., cells exposed to a concentration of a particular compound), four independent wells were simultaneously incubated with 3.75 ng/mL BODIPY 493/503, 1.5 µg/mL PI and 2 µg/mL Hoechst 33342 for 30 min, while four parallel wells in a twin 96-well plate were incubated for 30 min with a second mixture of probes (75 µg/mL TMRM, 2 µg/mL DHCF-DA, 1.5 µg/mL PI, and 1.5 µg/mL Hoechst 33342). After staining, samples were analyzed with Scan (Olympus), an HCS system based on automated epifluorescence microscopy, and an image analysis of cells in a microtiter plate format. The instrument enables fully automated monitoring of fluorescence intensity at four wavelengths, as well as image analysis and data viewing.
The 10× objective was used to collect images for the distinct fluorescence channels. To capture enough cells (>500) for the analysis, nine fields per well were imaged. Dyes were excited, and their fluorescence was monitored at the excitation and emission wavelengths with an appropriate filter set. The analysis was performed by using the Automated Image and Data Analysis Software (the OSIS Scan software), which not only allows the simultaneous quantification of subcellular inclusions that are marked by different fluorescent probes but also measures fluorescence intensity associated with the predefined nuclear and cytoplasmic compartments. Hoechst 33342 fluorescence was used to identify the nuclear region and was measured as the main object. To determine cell viability, the PI fluorescence intensity in the nuclear region was measured, and positive staining was considered to be indicative of death cells. The cellular mitochondrial membrane potential was defined as the TMRM fluorescence intensity in the punctate cytosolic regions around the nucleus. To assess intracellular fat deposits, BODIPY 493/503 fluorescence intensity was measured in a large intracellular region. ROS production was defined as the DCF fluorescence intensity in punctate cytosolic regions. Each measure was performed in individual cells and then averaged for the different treatments. Cells exposed to tested compounds but not incubated with dyes (blank wells) were used to correct potential interferences due to intrinsic fluorescence of compounds (i.e., doxycycline). The results obtained in the compound-treated cells were expressed as a percentage of the corresponding parameter (lipid content, ROS generation, viability, or mitochondrial membrane potential) in the solvent-treated cells (assumed as a 100% value).
Statistical Analysis
The data are expressed as the mean ± SD of at least three different experiments in quadruplicated wells. The Student t test was used to determine the statistical significance of the experimental data.
Results and Discussion
The present study aimed to develop a hepatocellular assay to be used as an in vitro screening tool to identify the drugs that cause steatosis early in the drug discovery and development pipeline. The assay consists of the application of a multichannel-fluorescence analysis using an HCS platform to determine specific cellular and biochemical changes associated with chemically induced steatosis. To this end, the HepG2 cells previously exposed to several concentrations of tested compounds were incubated with fluorescent dyes: BODIPY493/503 to determine lipid accumulation, DHCF-DA to measure ROS generation, TMRM as indicative of mitochondrial membrane potential changes, and PI to evaluate cell viability. These fluorescent probes were selected on the basis of the previous information obtained from a flow cytometry method developed in our laboratory.
6
HepG2 cells, despite having a lower drug-metabolizing capacity than primary hepatocytes, have proved to be a very useful hepatocellular model for the study of liver steatosis, particularly drug-induced hepatosteatosis.4,6,7 Moreover, comparable increases in lipid accumulation have been reported in HepG2 cells and hepatocytes exposed to steatosis inducers.
4
As an example of the potential utility of this assay,
Figure 1
shows images of untreated HepG2 cells and cells exposed for 24 h to tetracycline, a well-known steatogenic drug.3,9A concentration-dependent lipid overaccumulation was found, and the effect became evident at noncytotoxic tetracycline concentrations (200 µM;
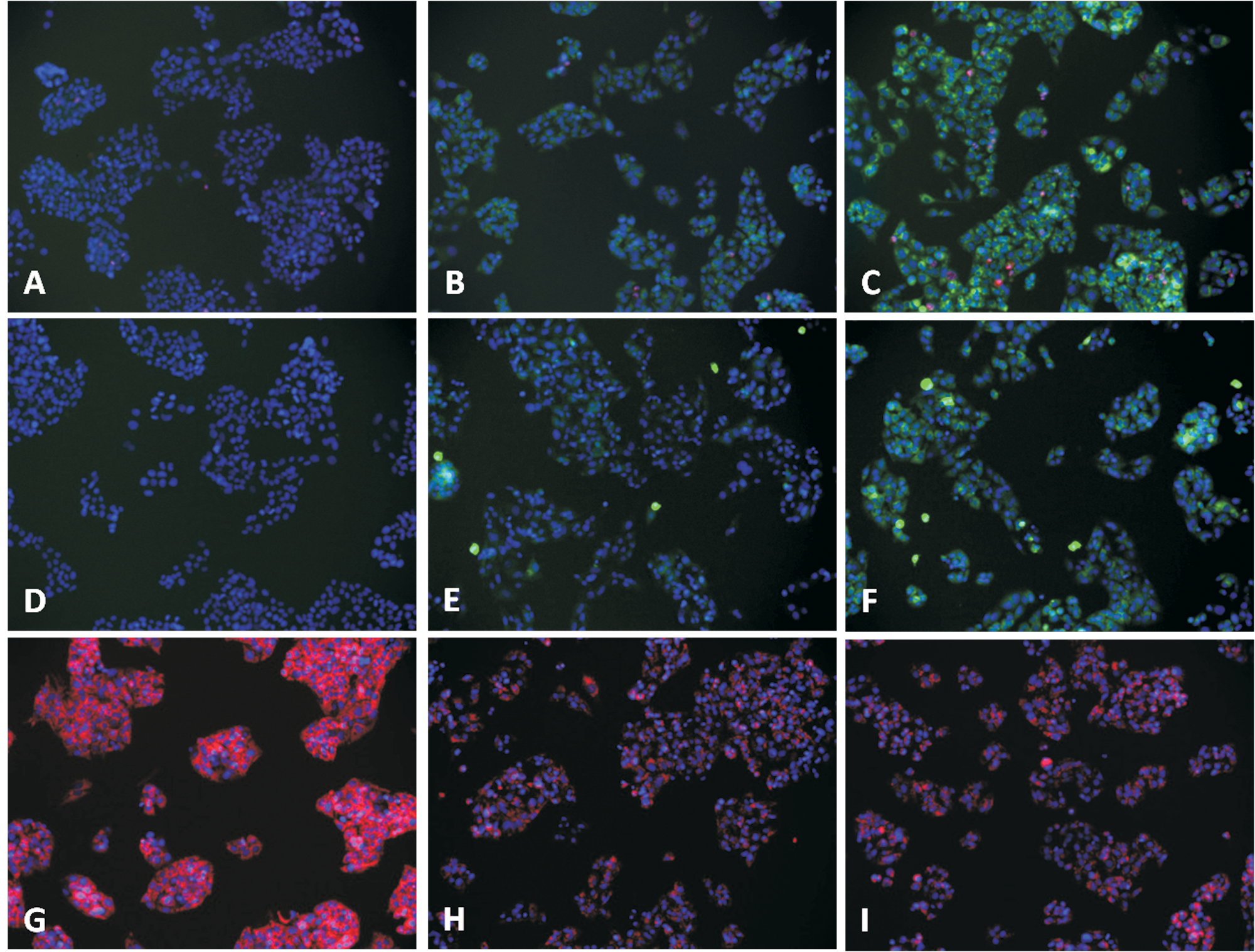
Representative images from the high-content screen for the prediction of the steatotic potential of drugs. Nontreated HepG2 cells (A, D, G) exposed to tetracycline 200 µM (B, E, H) or tetracycline 800 µM (C, F, I) are shown. Fluorescence of BODIPY493/503 (to stain neutral lipids; green), Hoechst 33342 (to detect nuclei; blue), and propidium iodide (to determine cell viability; red) (A, B, C) or fluorescence of 2′,7′-dichlorofluorescein (to detect reactive oxygen species production; green) and Hoechst 33342 (D, E, F) or of TMRM (to detect changes in mitochondrial membrane potential; red) and Hoeschst 33342 (G, H, I) is exemplified.
A total of 22 compounds harboring a variety of chemical structures and biological effects were tested using the proposed multiparametric assay. The list included 16 drugs reported to cause liver steatosis through different mechanisms (impairment of fatty acid β–oxidation enzymes, sequestration of CoA, damage to mitochondrial DNA with subsequent mitochondrial dysfunction, inhibition of lipoprotein secretion): acetylsalicylic acid, amiodarone, clofibrate, cyclosporine A, didanosine, doxycycline, DL-ethionine, fenofibrate, fialuridine, stavudine, tamoxifen, tetracycline, tianeptine, ticlopidine, valproate, and zidovudine (positive controls). In addition, six nonsteatotic compounds, but with variable toxic effects, were included in the study as negative controls: amikacin (phospholipidosis), rotenone (mitochondrial toxicity), t-butyl hydroperoxide and cumene hydroperoxide (oxidative stress), colchicine (oxidative stress, cell cycle disturbance), and citrate (nontoxic compound). Concentration-dependent results obtained after examining lipid accumulation, ROS generation, cell viability, and mitochondrial membrane potential in HepG2 cells treated with the tested compounds are shown in
Figure 2
and
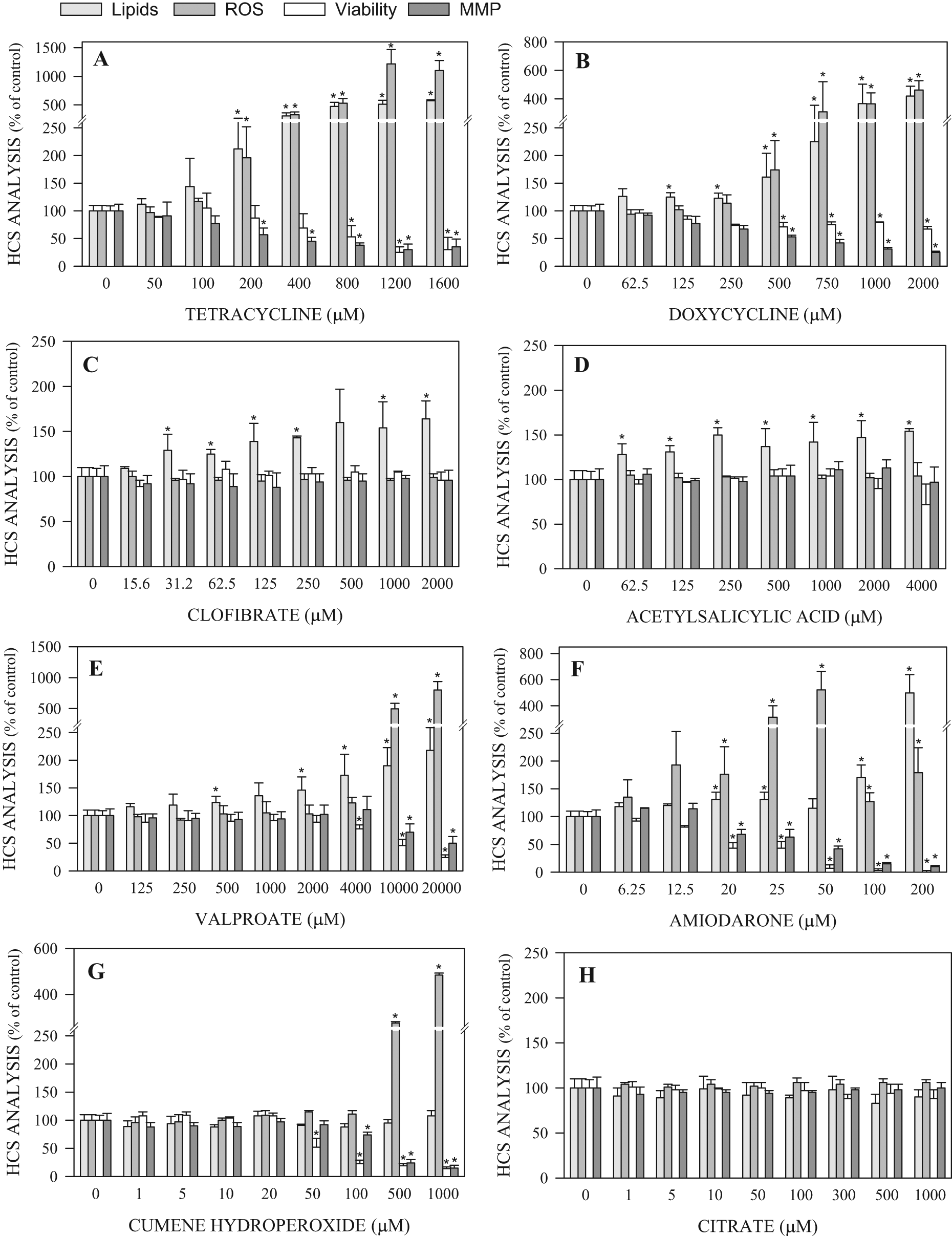
Multiparametric assessment of concentration-dependent effects of steatogenic compounds on HepG2 cells. After 24 h exposure of cultured cells to increasing concentrations of the compounds, lipid content (evaluated by BODIPY483/503 fluorescence), reactive oxygen species generation (evaluated by 2′,7′-dichlorofluorescein fluorescence), cell viability (evaluated by propidium iodide fluorescence), and mitochondrial membrane potential (evaluated by tetramethyl rhodamine methyl ester fluorescence) were analyzed by high-content screening. The results are expressed as a percentage of the control (solvent-treated cells) and correspond to the mean ± SD of at least 3 independent experiments. *p < 0.05.
Quantification of intracellular BODIPY493/503 fluorescence intensity revealed that all the drugs previously reported as steatosis inducers produced significant increases in fat deposits in the HepG2. The highest effects on lipid levels were induced by amiodarone, doxycycline, and tetracycline (up to about five-fold more than untreated cells;
Fig. 2
). For most steatosis-positive compounds, significant increases in intracellular lipid accumulation were produced at subcytotoxic concentrations of the drugs. In contrast, no effects on lipid content were observed after exposing HepG2 cells to the steatosis-negative chemicals, even at concentrations that led to significant effects on the other parameters analyzed (viability, ROS generation, or mitochondrial membrane potential;
In parallel to lipid overaccumulation, tetracycline and doxycycline produced a concentration-dependent increase in ROS generation and a depolarization of the mitochondrial membrane (
In contrast, other steatosis-positive compounds led to increased intracellular lipid accumulation with no remarkable changes in peroxidative activity. Among them, antiviral agents, including nucleoside reverse transcriptase inhibitors (didanosine, stavudine, zidovudine) and filauridine, have been reported to induce microvesicular steatosis by causing mitochondrial DNA depletion.1,2 Accordingly, the evaluation of their effects on HepG2 cells revealed changes in lipid content and mitochondrial membrane potential but no effects on ROS formation, and fialuridine displayed the highest toxicity (
Our results are in agreement with a previous study using a gene expression analysis of hepatocytes exposed to noncytotoxic concentrations of positive steatosis reference molecules, which revealed two different groups of compounds: those that upregulated oxidative stress genes (i.e., tetracycline) and those that did not change oxidative stress genes (i.e., clofibrate). 5 High ROS levels in a lipid-enriched environment can promote lipid peroxidation, leading to aldehydic derivatives with detrimental effects on hepatocytes, which may account for the steatohepatitis lesions induced by drugs. 2 Therefore, risk of liver damage by a steatotic mechanism is likely to be higher for those drugs that increase lipid accumulation and ROS generation in a combined manner (i.e., amiodarone, cyclosporine A, doxycycline, tamoxifen, tetracycline, or valproate).
The present assay could be helpful to detect potential steatosis inducers at early stages of drug development. In vitro data should be interpreted as a preliminary indication of the steatogenic potential of a new compound but not as a direct index of their in vivo effects. Pathological action of a particular steatotic drug depends not only on the ability of the compound to increase intracellular lipid content (estimated from in vitro assays) but also on the administered dose and the duration of the treatment that determine serum levels and potential hepatic accumulation of the drug.
In summary, the present fluorescent-based approach seems to be a promising screening tool for steatosis-inducing compounds and offers certain advantages over other previously reported in vitro assays.3–6,15 One major characteristic of the assay is its relative simplicity when compared with more complex or time-consuming methods based on genomic, proteomic, or flow cytometry analyses.5,6,15 Moreover, the use of 96-well formats increases assay high throughput, thus enabling the comparative testing of large sets of compounds in the same experiment. The assay correctly identified positive and negative steatosis inducers, independently of their chemical and biological properties (i.e., compounds that produce fat overaccumulation through different mechanisms or those showing other hepatotoxic effects). Other features of the method include its sensitivity, which allows the testing of low noncytotoxic concentrations of the compounds; the use of hepatic cells of a human origin, thus avoiding interspecies extrapolations; the combination of several probes indicative of specific mechanisms of toxicity; and the identification of early cellular events with potential chronic consequences. To confirm the utility of the present in vitro assay for the screening of the steatogenic potential of new drugs, a larger number of compounds with contrasted in vivo information should be tested.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work has been supported by the Instituto de Salud Carlos III of Spanish Ministry of Health (grants IF08/3638, PI10/00923).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.