
Abstract
Pooled short-hairpin RNA (shRNA) library screening is a powerful tool for identifying a set of genes in biological pathways that require stable expression to produce a desired phenotype. Massive parallel sequencing of half-hairpins has proven highly variable and has not given satisfactory results concerning the relative abundance of different shRNAs before and after selection. Here, the authors describe a method for quantitative comparison of half-hairpins from pooled shRNAs in the mir30-based pGIPZ vector that is analyzed by massive parallel sequencing. Introducing a multiplexing code and refining the sample preparation scheme resulted in the predicted ability to detect twofold enrichments. These improvements should permit half-hairpin sequencing to analyze either dropout screens or selective pooled shRNA screens of limited stringency to analyze phenotypes not accessible in transient experiments.
Keywords
Introduction
Short-hairpin RNAi (shRNA) libraries and the ability to perform large-scale gain or loss-of-function screening are powerful tools for identifying new sets of genes involved in biological pathways.1,2 High-throughput one-well/one-target screening using transient small interfering RNA (siRNA) oligonucleotides or shRNAs have been widely used for phenotypes that produce rapid effects (e.g., Cerone et al. 3 and Lamarcq et al. 4 ). However, it is impractical to perform one-by-one assays for phenotypes that require a longer time to be expressed. Stable shRNA integration of pooled shRNAs provides a unique advantage for these long-term phenotypes, which cannot be assayed in the few days of a high-throughput assay. Pooled shRNAs can be identified by first recovering the integrated sequences from cells with and without selection and then determining whether their abundance has changed with respect to total shRNAs by either half-hairpin/barcode microarray5-7 or massive parallel sequencing of half-hairpins to transform the biological screening result into quantitative and comparable data ( Fig. 1A ).
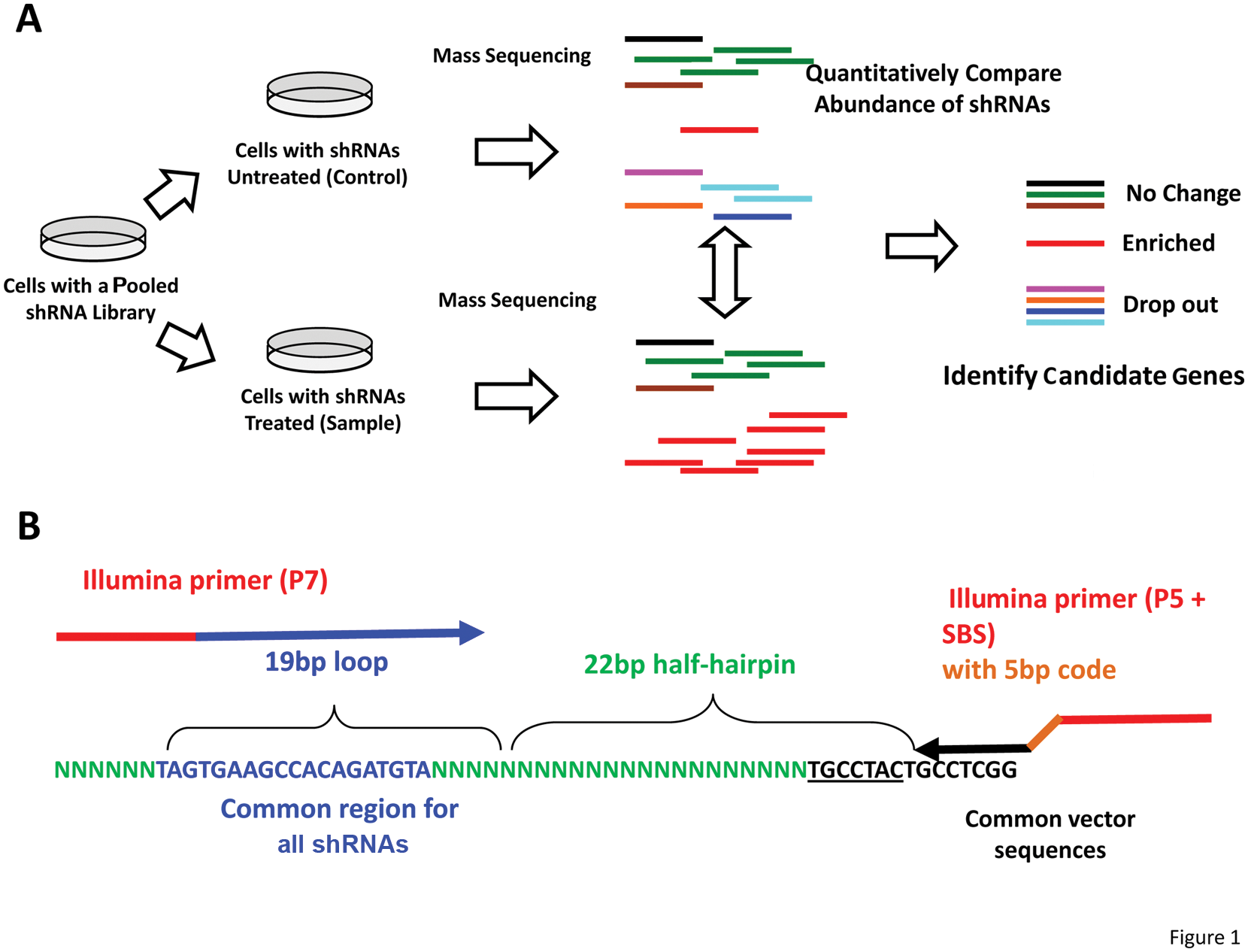
Analysis of pooled short-hairpin RNA (shRNA) library screening using massive parallel sequencing. (
We initially developed a method for amplifying half-hairpins from a 62 000–member mir30-based pGIPZ shRNA library (Open Biosystems, Huntsville, AL) that could be analyzed by deep sequencing ( Fig. 1B ). By introducing a multiplexing code, we could decompose the variability introduced by PCR reactions and determine the minimum number of samples and reads needed to obtain statistically meaningful results for different fold-enrichments following selection. These initial results suggested there would be significant difficulties in reliably detecting fourfold enrichments. We thus made a smaller custom 13 530–member shRNA library using oligonucleotides synthesized using microarray printing technology, refined the PCR amplification scheme, and repeated the analysis. These improvements resulted in the predicted ability to detect twofold enrichments. Below, we describe these improvements that should now permit half-hairpin sequencing to be able to analyze either dropout screens or selective shRNA screens of limited stringency (where the difference after selection is not all-or-none, as in growth in soft agar, but a limited enrichment of several-fold) to analyze phenotypes not accessible in transient experiments.
Materials and Methods
Construction of a custom shRNA library with emulsion PCR
We selected 451 genes known to be involved in any aspect of telomere biology/telomerase or DNA damage pathways as part of other experiments. Thirty shRNAs for each gene were designed using the RNAi Central algorism (http://katahdin.cshl.org/siRNA/RNAi.cgi?type=shRNA) as used in Bassik et al. 8 The output sequences for 97-mers of these 451 genes were formatted in an Excel sheet and sent to MYcroarray.com at Ann Arbor, Michigan, where they were digitally synthesized with their proprietary microarray fabrication technology, cleaved from the glass substrate, and returned as a pool in solution. Gel analysis showed a smear of products rather than a 97-mer band. PCR comparing the ability to amplify this mixture versus a conventionally synthesized 97-mer suggested that ~1:1000 of the oligonucleotides were amplifiable. We then amplified the library using emulsion PCR to minimize distortion during amplification, as described in Williams et al., 9 using the more commonly available oil–surfactant mix made of Span-80, Tween-80, and Triton X-100 with KOD Hot Start Polymerase (Novagen, Rockland, MA). Emulsion PCR premix was prepared with the forward primer (XhoI site underlined 5′-TTCTCGAGAAGGTATATTGCTGTTGA CAGTGAGCG-3′ and the reverse primer (EcoRI site underlined) 5′-TTGAATTCCGAGGCAGTAGGCA-3′. Each primer was used at a final concentration 0.3 µM and with 0.05 U/µL KOD Hot Start polymerase and with a template input of 1 pmol of the oligo library. Then, 260 µL of the aqueous premix was added dropwise into 400 µL of the mineral oil/detergent mixture while stirring at room temperature at 1000 rpm for 10 min, then divided into eight microfuge tubes. PCR was performed at 25 cycles of 95 °C for 30 s, 65 °C for 30 s, and 72 °C for 45 s. Emulsion PCR reactions were recovered by breaking the emulsion by centrifugation at maximum speed for 10 min and organic extraction with diethyl ether. The emulsion PCR products were combined at this step and digested with EcoRI and XhoI for cloning into the lentiviral shRNAmir pGIPZ vector.
Sequencing primers were added to the PCR products prior to cloning and the distribution of different shRNAs determined by deep sequencing, comparing the efficiency of normal versus emulsion PCR in minimizing variability within the library. Emulsion PCR yielded a lower coefficient of variation than normal PCR (
The pGIPZ shRNAmir vector has two EcoRI sites. We first destroyed the site not used for shRNA cloning by partial digestion with EcoRI and self-ligation. After successful removal of one EcoRI site, the pGIPZ vector was digested with EcoRI and XhoI, and the appropriate plasmid band was gel purified.
Ligation was performed with 500 ng of the vector and 30 ng of the Xho1–EcoR1 digested/gel-purified emulsion PCR product using 500 U of T4 ligase in a total volume of 25 µL at 16 °C overnight. Then, 4 µL of the ligation reaction was transformed in 40 µL of SURE electroporation competent cells (Stratagene, Santa Clara, CA) at 25 µF capacitance, 200 Ω resistance, and 17 kV/cm field strength voltage with the Bio-Rad Gene-Pulsar (Bio-Rad, Hercules, CA). After electroporation, competent cells were suspended in 1 mL of LB media and incubated for 1 h at 37 °C, and the entire transformation was plated on a predried 150-cm ampicillin bacterial plate. The number of colonies recovered from each transformation was at least 30 000. The transformation was repeated 45 times to generate approximately 1.4 million clones (100-fold representation of each of ~13 530 shRNAs). Colonies on plates were harvested by scraping and directly used for DNA purification (Maxi-prep; Qiagen, Valencia, CA) without further amplification. We obtained ~1 mg of this cloned pGIPZ shRNA library.
Sample preparation of mir30 shRNAs for sequencing (Fig. 2)
Step 1: Enrichment of samples by biotin-streptavidin bead pull-down
107 polyploidy cells contain ~100 µg of genomic DNA. If infected at a low multiplicity of infection (MOI), so that the majority of cells received only one shRNA, there would be only 107 molecules of shRNA (= 16.6 × 10–18 mol = 16.6 attomol) in 100 µg of genomic DNA. An shRNA with an abundance of 4 per million would thus be present in only 40 copies per 100 µg. To recover this low abundance of these pooled shRNAs, isolated genomic DNA (100 µg or more) was fragmented using a Misonix 3000 cup horn sonicator and digested with XhoI and ApaLI overnight. The DNA was melted in 2× saline-sodium citrate (SSC) and then annealed in solution to 100 nM of a biotinylated locked nucleic acid (LNA) pull-down primer designed to a common region of the pGIPZ vector, ~400 bp downstream of the shRNA region internal to the ApaL1 site (5′-BioTEG-CTATTGCTTCCCGTATGGCT-3′, where the underlined bases are LNA). The solution was heated at 95 °C for 3 min and then cooled to 20 °C with a ramp of 0.1 °C/s. Then, 20-µL MyOne C1 beads (Dynal, Carlsbad, CA) were washed with 80 µL binding solution from the Dynal KilobaseBINDER Kit and then resuspended with 50 µL binding solution and 10 µL dH2O. The annealed DNA was mixed with the beads and rotated at room temperature (RT) for 2.5 h at 6.5 rpm. After washing once with 100 µL washing solution, the beads were resuspended in 50 µL Tris-EDTA (TE) and heated at 85 °C for 10 min to release the bound DNA and quickly placed on ice for 1 min. The eluted hairpins were collected in a separate tube.
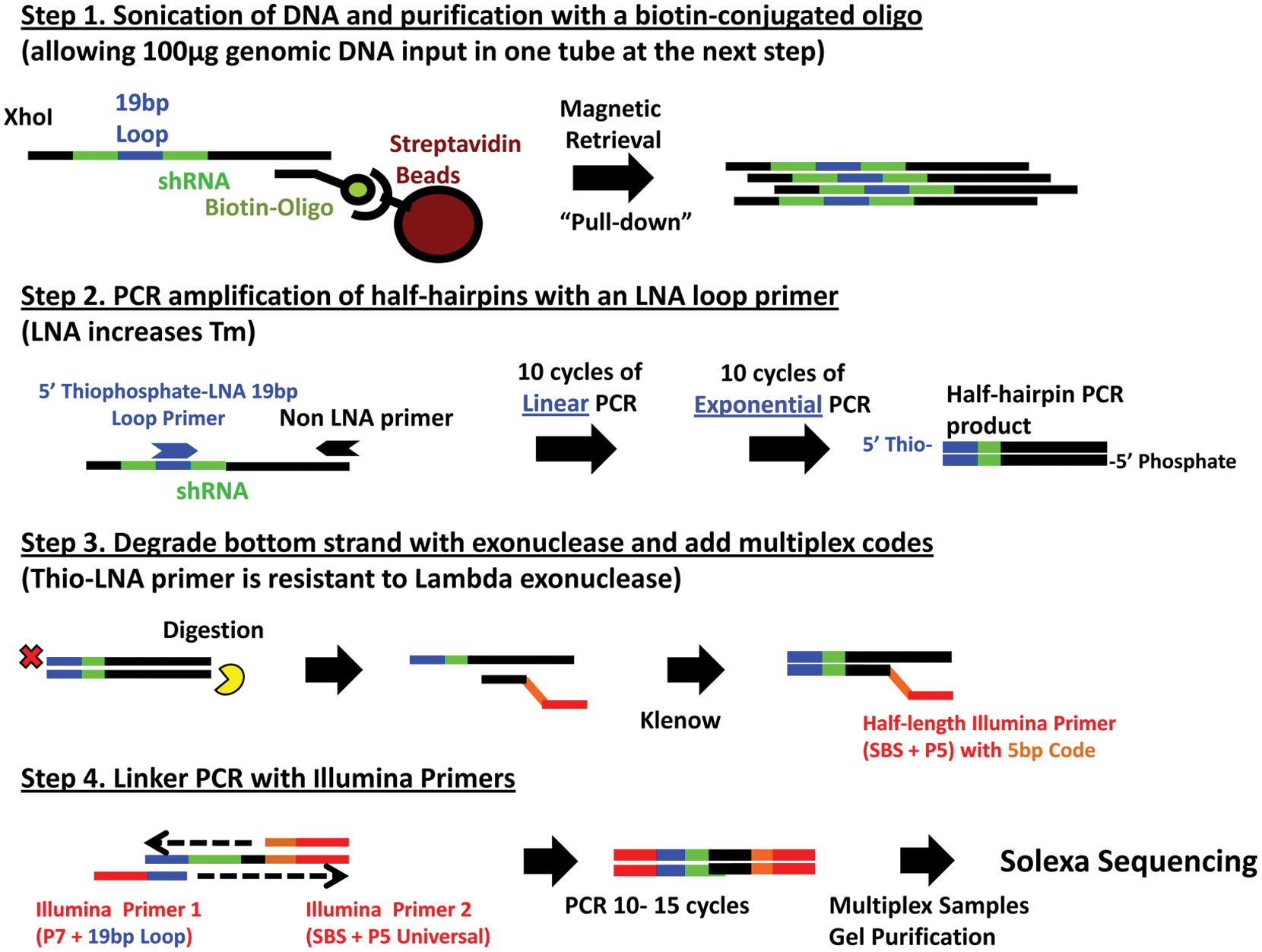
Optimization of half-hairpin sample preparation. Step 1: Retrieval of the low-abundance pooled short-hairpin RNAs (shRNAs) allows targets from 100 µg of genomic DNA to be used as input in one tube at the next PCR step. Step 2: Uniform conversion of full-hairpins to half-hairpins is a critical step to conserve the original distribution of the pooled shRNAs. Linear amplification is first performed to minimize conversion bias, which is then followed by normal PCR. Step 3: Lambda exonuclease degrades one strand but is unable to degrade 5′-thiophosphate-modified DNA. This prevents reannealing, which then allows the long low-temperature incubations needed for annealing of the multiplexing primer and extension by Klenow. Step 4: Minimize number of PCR cycles. Test PCR is performed to determine the minimal number of cycles needed to see a product. After amplification, multiplexed samples are combined prior to gel purification. See Materials and Methods for a detailed description of each step. LNA, locked nucleic acid; Tm, temperature.
Step 2: Conversion of hairpins to half-hairpins with LNA loop primer
To limit PCR bias during the full- to half-hairpin conversion and to increase the amount of each template before separating into four independent PCR reactions for multiplexed replicates, the eluted enriched shRNAs were linearly (rather than exponentially) amplified with the forward LNA loop primer, targeting the 19-bp common region of the shRNA hairpin. Ten cycles of 95 °C for 30 s, 63 °C for 30 s, and 72 °C for 45 s were performed using the KOD Hot Start Polymerase Kit (Novagen) with only the forward LNA primer (5′-TAGTGAAGCCACAGATGTA-3′, where the first four bases have phosphorothioate linkages and where the underlined bases are modified as LNA [Bio-Synthesis, Lewisville, TX]) using the shRNAs eluted from the pull-down. We found that amplification was inhibited if any binding/washing solution was present from the previous pull-down. This can be avoided by careful removal of binding and washing solution before eluting the shRNAs and by simply diluting the elution with 1× TE and separating it into at least four tubes for the linear PCR amplification. The 10-cycle linearly amplified products can then be recombined and divided into four replicate tubes for the next step of exponential PCR that includes the reverse primer. Ten cycles of 95 °C for 30 s, 63 °C for 30 s, and 72 °C for 45 s were performed after adding 0.3 µM of the reverse primer (5′-GAGTAATGGCCGGCCGCATTAGT-3′). The primers were then removed by the MiniElute PCR purification kit (Qiagen). At the end of this step, the pooled molecules would be at most 1.0 × 107 × 10 (linear) × 210 (exponential) = 1.0 × 10>11 molecules (0.166 picomol), which is still too low to be able to see on a gel.
Step 3: Strand-specific degradation and Klenow addition of 5-bp multiplexing codes
The 5′ LNA primers contained thiophosphate linkages to make them resistant to lambda exonuclease. The bottom strand of each PCR product was degraded using 5 U lambda exonuclease (NEB, Ipswich, MA) in 1× KOD buffer at 37 °C for 1 h followed by heat inactivation at 75 °C for 10 min, leaving the top strand intact. A P5–SBS–half-primer with a 5-bp unique code (
Step 4: Linker PCR with Illumina primers
The P7-loop and P5-SBS-Universal primers (
Acquisition of data
We usually obtained at least 20 million raw sequences. They were first sorted according to the 5-bp multiplex code, and then the half-hairpin reads were matched to our reference shRNA sequence file using the E-land aligner. In this script, we allowed two mismatches within the 22-bp half-hairpin region, but no mismatch was allowed in the 5-bp multiplex code region. The acquired sequencing data were first normalized by upper quartile normalization and then transformed into a Log2(X + 1) scale.
Virus packaging and transduction
A DNA pool of the 13 530 lentiviral custom shRNA library or the 62 000 shRNA library (Open Biosystems) was co-transfected with packaging plasmids PAX2 and pMD2 in 293FT cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The filtered viral supernatant was then transduced into VA13-derived cells with Polybrene.
Results and Discussion
Sample preparation is the major source of variability in sequencing data
We initially encountered large variability in the abundance of individual shRNA sequences even when using identical starting DNA. To identify the source of this variability, we independently prepared six sequencing samples (experimental replicates) from a single genomic DNA sample from cells transduced with a pooled 62 000 shRNA library (Open Biosystems). One of the six samples was analyzed in one entire lane of an Illumina (San Diego, CA) GAIIX (single lane), and the other five samples were multiplexed and combined in a second lane with many fewer reads per sample (multiplexed) (
Optimization of half-hairpin sample preparation
We have optimized four critical steps in sample preparation that have collectively dramatically reduced the variation between samples (
Fig. 2
). Tens to hundreds of micrograms of DNA are required to maintain the complexity of the initial input, but more than 1 to 2 µg of total DNA is inhibitory to the initial PCR reactions. The first step is thus to eliminate the bulk of total DNA by selectively retrieving the integrated shRNA regions by first hybridizing the denatured DNA to a biotin-containing oligonucleotide complementary to the lentiviral sequences common to all the shRNAs and then recovering them using streptavidin-coated magnet beads (
Fig. 2
, step 1).
Step 2 is designed to convert the full-hairpin shRNAs into half-hairpins while minimizing any sequence-specific bias between different hairpins ( Fig. 2 , step 2). Two modifications were introduced to accomplish this. Half-hairpins are produced using a primer (to the common 19-bp loop region containing three LNA substitutions) that increases the temperature (Tm) from 48 °C to 63 °C. The increased Tm allows higher annealing temperatures and thus decreases the stability of snap-back hairpin formation from those sequences that might inhibit the production of half-hairpins. Bias in conversion to half-hairpins was also reduced by initially performing linear PCR. Bias then increases in only a linear fashion, so that after 10 cycles, a hairpin with only 10% conversion efficiency has nonetheless been converted to 10% of the half-hairpins rather than perhaps the 1/1000 that could occur during exponential amplification. After 10 cycles of linear PCR with just the 19-bp loop-primer, the reverse primer is added for the initial exponential amplification of the minimally biased input of half-hairpins.
The third step adds both a multiplex code and the PCR sequences needed for Solexa sequencing (
Fig. 2
, step 3). These sequences needed to be introduced within the 36-bp read length of relatively accurate sequencing, and thus we limited the overlap of sequences needed to introduce these changes to the seven vector nucleotides immediately adjacent to the half-hairpins. Preliminary experiments demonstrated that reannealing of upper and lower strands inhibited the efficiency of this step under the long annealing/extension conditions that otherwise maximized its efficiency. We thus introduced a second modification into the loop-primer that allowed us to produce a single-stranded template for this step. The 5′ to 3′ lambda exonuclease is unable to digest ends containing several 5′-thiophosphate linkages. The initial PCR was thus performed with a 19-bp loop primer that contained both 5-thiophosphates and LNA substitutions. Treatment of the PCR product with lambda exonuclease then selectively degraded the bottom strand, leaving a single-stranded upper strand template for the introduction of the multiplex codes and sequencing primers using Klenow (
The last step is to perform PCR with modified Illumina primers that add the remaining nucleotides needed for sequencing and determining the lowest number of PCR cycles needed to faintly visualize the product to minimize general PCR bias. Equal amounts of DNA from different multiplexed samples are then combined, gel purified, and sequenced.
Refined method decreased experimental variability and increased sensitivity
To test if these modifications in sample preparation could reduce the variability among experimental replicates, we independently prepared four sequencing samples from a single sample of genomic DNA from cells transduced with a pooled 13 530–member custom library of mir30-based pGIPZ shRNAs ( Fig. 3A ; see Materials and Methods for construction of the custom shRNA library). The enriched pull-down elution was amplified by 10 cycles of linear amplification with the thiophosphate–LNA primer to minimize both half-hairpin conversion bias and input variation prior to separating into four samples to avoid sampling problems. The four samples were prepared as described above, multiplexed, and sequenced together in a single lane along with other multiplexed samples. Sequencing data generated ~1 million reads per sample, and each set of data was normalized and transformed to a Log2(X + 1) scale. The number of sequencing reads (abundance) per individual shRNA in each sample was plotted against its abundance in each of the other three samples ( Fig. 3B ). These data showed good consistency among replicates, although there are some outliers in each comparison. The modified method dramatically decreased the overall coefficients of variance across replicates ( Fig. 3C ). Using these modifications, about 90% of 13 530 shRNAs have a coefficient of variance below 0.3, and about 80% of shRNAs have a coefficient of variance below 0.2. The low coefficient of variance enables the experiment to detect subtle changes in shRNA abundance. Figure 3D shows how the detectable fold change for 90% of the custom shRNAs varies with different numbers of replicates based on our data. It is derived by assuming experimental conditions and quality are fixed, with only the number of replicates changing. Figure 3E summarizes how the coefficients of variance and number of replicates determine the detectable fold changes, as well as the correspondence between the coefficient of variance and the number of shRNAs that fall within these limits in our experiments. On the basis of these results, we selected four replicates as representing a reasonable balance between practicality and increased accuracy.
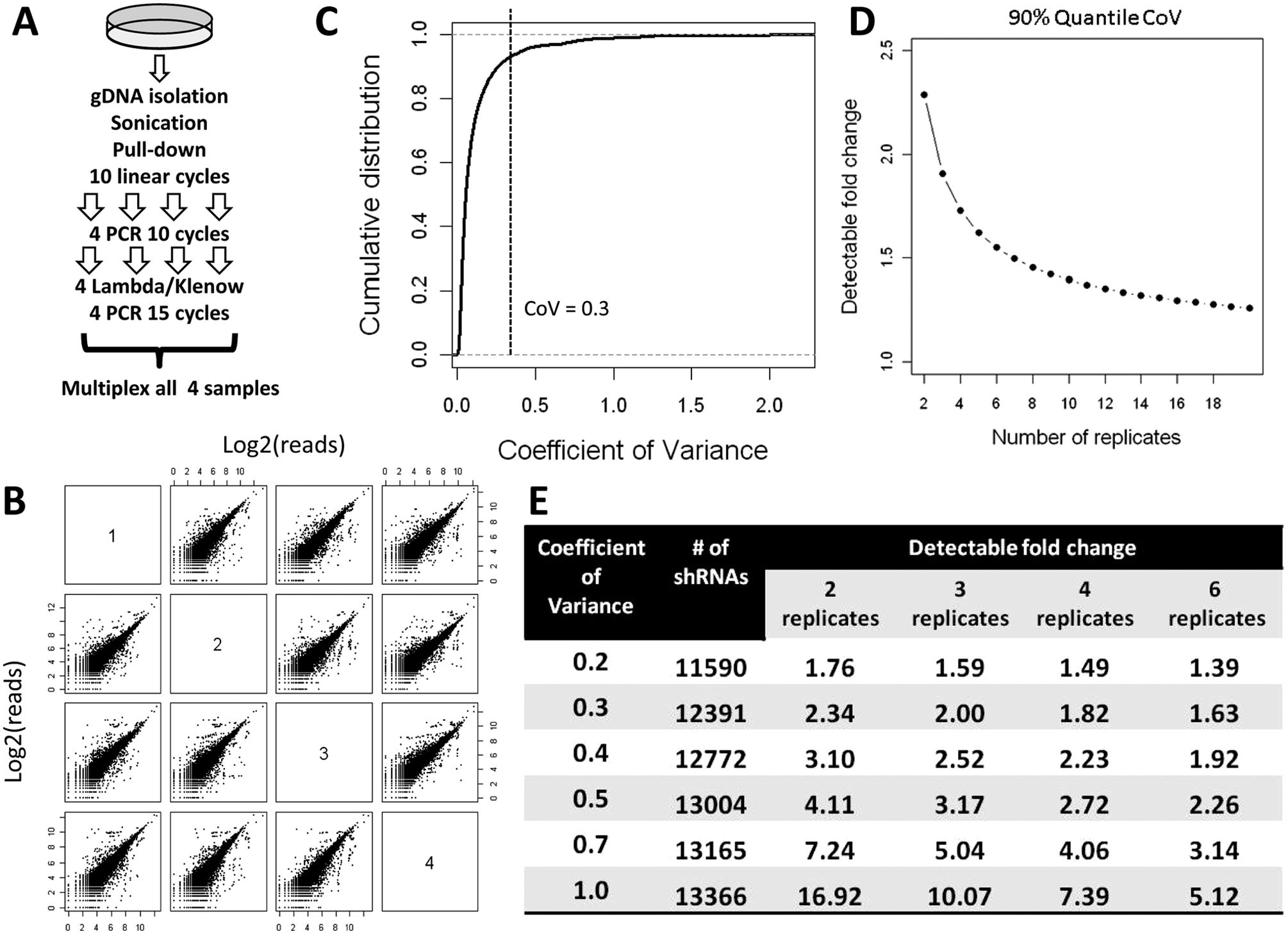
Refined method decreases experimental variability and increases the detection of small fold changes. (
Analysis of the refined method by means of spiked-in shRNAs
We further validated the refined sample preparation by spiking the genomic DNA with defined amounts of 25 reference pGIPZ shRNA plasmids that were not present in the custom shRNA library. Each spiked shRNA was carefully diluted to 20 molecules/µL and mixed with the genomic DNA transduced with the custom shRNA library before pull-down to see if the small amount of reference shRNA plasmids were represented at the appropriate abundance among repeated experiments (biological replicates). Sequencing results showed that measured shRNA ratios were accurate compared with the designed ratios, and the results were consistent among the experiments ( Fig. 4 ). This method is thus reliably recovering even low-abundance reference shRNAs and accurately detecting fold changes between samples.
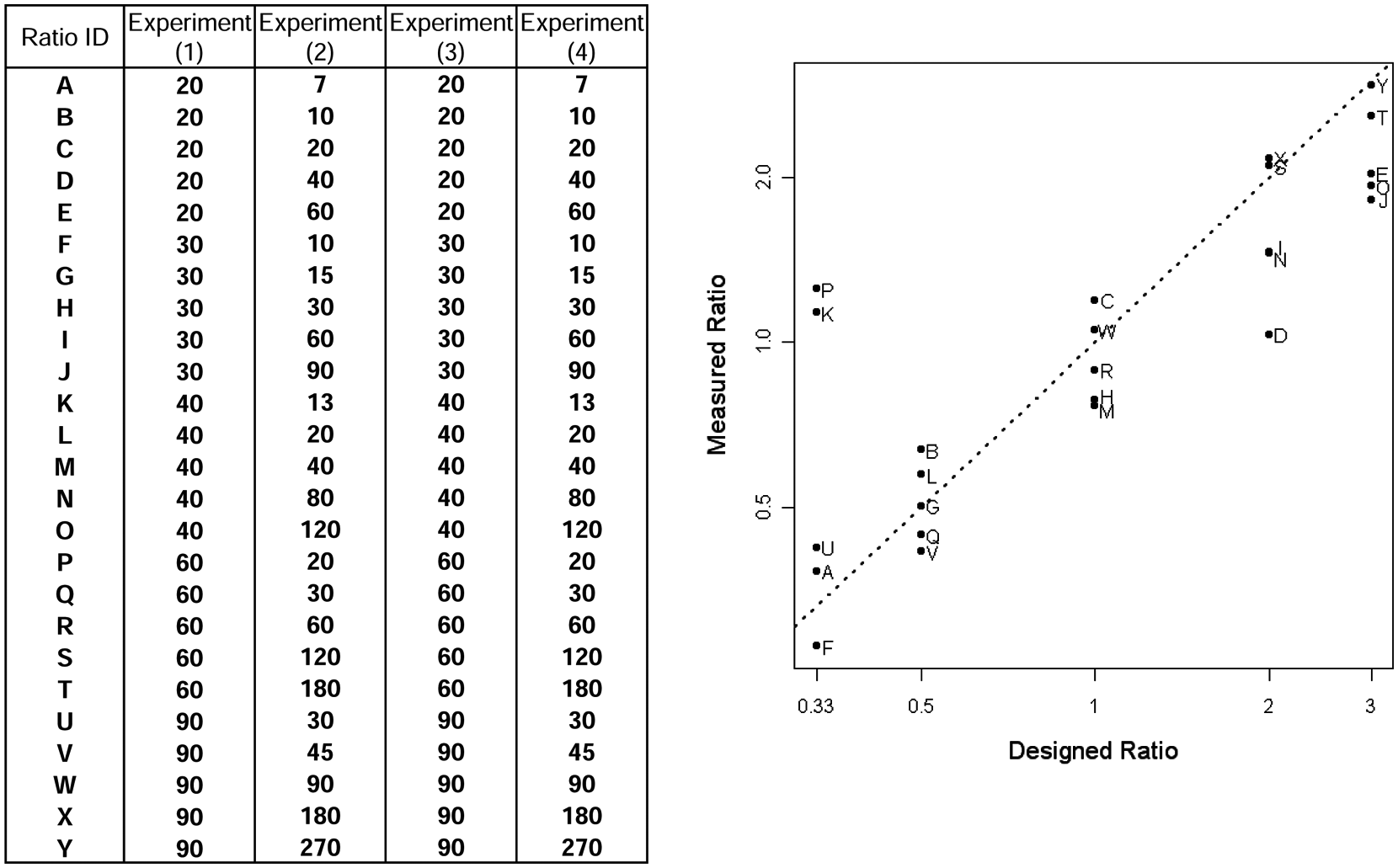
Validation using spiked-in short-hairpin RNAs (shRNAs). Twenty-five reference shRNAs that do not exist in our custom shRNA library were diluted to 20 molecules/µL. Inaccuracies in determining the initial concentration and during serial dilutions will produce some variation. Combinations of these diluted pGIPZ plasmids were mixed with genomic DNA transduced with the custom shRNA library as shown prior to the pull-down step. Four multiplexed replicates were made for each of the four experiments. Experiments 1 and 3 are duplicates of what could be considered the abundance prior to selection and experiments 2 and 4 the abundance following selection. The x-axis is the designed ratio (fold change) between samples (mean reads of experiments 1 and 3/mean reads of experiments 2 and 4). The y-axis is the observed fold change based on sequencing data. The figure confirms that the measured fold change between samples in most cases accurately reflects the designed ratio for shRNAs with sufficient abundance.
The refined method of sample preparation described here demonstrates significant improvements for sequencing shRNA screens in both data quality and the resulting ability to detect small fold changes in abundance (
Microarray analysis is currently the favored method for the analysis of large-scale pooled shRNA library screening. Microarray analysis using long barcodes is only suitable for commercial libraries and is not readily adapted to custom libraries. Microarray probe preparation requires large amounts of PCR product for competitive hybridization of fluorescence-labeled probes (half-hairpins or barcode sequences), which could contribute PCR bias. For half-hairpin microarrays, there will be complications due to weak and selective hybridization of the short ~22-bp variable-sequence probes. Furthermore, it is questionable whether detecting twofold change by the relative signal intensity of the two fluorescent probes is valid for anything but the more abundant shRNAs.
We have developed a method for a successful construction of high-complexity custom shRNA libraries targeting a pathway of interest and a method for the quantitative comparison of the pooled shRNA library by successfully recovering the half-hairpins and analyzing them using mass parallel sequencing. This allows one to design a pooled shRNA library targeting particular pathways that can be analyzed by deep sequencing of half-hairpins without relying on custom barcode microarrays, which are not available for custom libraries.
Footnotes
Acknowledgements
This work was supported by the National Institutes of Health (5R01CA152301 to Y.X., 1R21DA027592 to G.X.), the National Science Foundation (DMS-0907562 to G.X.), the Simmons Comprehensive Cancer Center, and the Texas Advanced Research Program (010019-0084-2007 to W.E.W.).
J.M.R. is co-founder and employed by Biodiscovery, LLC (DBA MYcroarray).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.