
Abstract
Triglyceride lipases such as lipoprotein lipase, endothelial lipase, and hepatic lipase play key roles in controlling the levels of plasma lipoprotein. Accordingly, small-molecule modulation of these species could alter patient lipid profiles with corresponding health effects. Screening of these enzymes for small-molecule therapeutics has historically involved the use of lipid-based particles to mimic native substrates. However, particle-based artifacts can complicate the discovery of therapeutic molecules. As a simplifying solution, the authors sought to develop an approach involving a soluble and monomeric lipase substrate. Using purified bovine lipoprotein lipase as a model system, they show that the hydrolysis of resorufin butyrate can be fluorescently monitored to give a robust assay (Z′ > 0.8). Critically, using parallel approaches, they show that resorufin butyrate is soluble and monomeric under assay conditions. The presented assay should be useful as a simple and inexpensive primary or secondary screen for the discovery of therapeutic lipase modulators.
Introduction
Endothelial lipase (EL), hepatic lipase (HL), and lipoprotein lipase (LPL) belong to the triglyceride lipase family and play key roles in regulating circulating lipoprotein levels. 1 These homodimeric enzymes are anchored to the luminal surface of endothelial cells—a localization historically attributed to their heparin sulfate proteoglycan binding activity (as reviewed by Bishop et al. 2 ), although this concept has recently been challenged. 3 EL, HL, and LPL share significant sequence similarity 4 and comprise two domains, an arrangement inferred through homology to pancreatic lipase—a closely related enzyme with a known 3D structure. 5 The N-terminal domain contains a serine esterase catalytic center that functions to hydrolyze phospholipids and triglycerides at the sn-1 position, 1 albeit with markedly different specificities. In particular, whereas LPL and HL preferentially hydrolyze triglycerides over phospholipids (at ratios of ~140:1 and ~24:1, respectively), EL hydrolyzes phospholipids at a marginally higher rate than triglycerides. 6 These specificities are imparted by sequence differences within the lid subdomain—a flap-like structure that forms part of the substrate binding site. 4
Modulation of EL, HL, and LPL should alter the balance of plasma lipoproteins and have corresponding effects on the progression of atherosclerosis.1,3 The pursuit of pharmacological modulators of lipases has traditionally involved particle-based substrates, including the use of isolated lipoproteins.7,8 This approach has the advantage of approximating native substrates; however, these systems complicate interpretation of experimental data and may produce assay artifacts. Specifically, interfacial substrate systems such as micelles, liposomes, and emulsions do not conform to Michealis-Menten kinetics since factors such as the following can affect reaction progression: (1) the association/dissociation of enzyme molecules with the surface of the particle-based substrate and (2) the alteration of particle morphology and composition as substrate is turned over. 9 Furthermore, compounds that influence these processes have been found to give false positives. 10 Similarly, we speculate that if the assay readout is dependent on particle morphology or integrity, compounds that alter these properties could result in false positives or negatives. As an alternative strategy, we sought to develop an assay involving a particle-free substrate. We elected to focus on resorufin butyrate for several reasons. First, the low logP value of this compound (1.57, calculated with the online tool jlogP) suggested that it should be water soluble with the potential to be monomeric in the desired concentration range (nanomolar to micromolar). Also, the processing of this substrate can be conveniently followed since one of the products (resorufin) is highly fluorescent ( Fig. 1 ). Furthermore, excitation and emission wavelengths used to follow the hydrolysis of resorufin butyrate (500 and 593 nm, respectively) are values that are outside the typical absorption/emission range of drug-like molecules, 11 thus minimizing interfering artifacts such as auto-fluorescence and fluorescence quenching from small-molecule library members. Finally, the small size of the molecule suggested that it should be able to penetrate the active site of triglyceride lipases. Indeed, resorufin butyrate is hydrolyzed by a diverse set of serine hydrolases, including cholinesterase and several lipases.12,13 Here, we build on this latter finding in several ways: (1) We show that the utility of resorufin butyrate can be extended to a triglyceride lipase family member (namely, LPL); (2) we define conditions where the substrate is soluble and monomeric, thus enabling a particle-free lipase assay; and (3) we develop and validate a high-throughput assay.

Resorufin butyrate can serve as a substrate for lipoprotein lipase. (
To expedite the investigation of the suitability of resorufin butyrate as a triglyceride lipase substrate, we chose to use commercially available bovine LPL (bLPL) as a model system. This enzyme was selected since it is readily available in a purified form, obviating the need for reagent production. With this system, we show that resorufin butyrate can be used as a triglyceride lipase substrate and adapt it for use in a robust, high-throughput assay. Critical to the goals outlined here, we show that resorufin butyrate is soluble and monomeric under the investigated conditions.
Materials and Methods
Chemical reagents
bLPL and resorufin butyrate were purchased from Sigma (Oakville, Ontario, Canada). The bLPL enzyme was formulated by the manufacturer as a suspension in 3.8 M ammonium sulfate, 0.02 M Tris HCl, pH 8.0 at 0.62 mg/mL (1.071 mM). For long-term storage, the enzyme was aliquoted, flash frozen, and stored at –80 °C. The working solution could be stored at 4 °C for several weeks without affecting activity. On the day of the assay, bLPL aliquots were freshly diluted into phosphate-buffered saline (PBS) and stored on ice until addition to the assay plate. M-352, a triglyceride lipase inhibitor, is a Merck proprietary sulfonyl fluoride compound that was synthesized at Merck Frosst Canada (Kirkland, Quebec). All other chemical reagents were purchased commercially at the highest quality available. All chemical compounds, including resorufin butyrate, were dissolved in DMSO and stored at –20 °C.
Lipase assay
The final assay volume was 50 µL in a 384-well plate. Enzyme stocks were diluted with PBS followed by the addition of either DMSO-solubilized inhibitor or pure DMSO (final assay DMSO concentration was 5%). Inhibitors/vehicle were added to a dry plate using an Echo 555 acoustic liquid handler (Labcyte, Sunnyvale, CA); subsequently, enzyme solution was added to the plate and sealed. Sealed plates were incubated at room temperature for 30 min with shaking prior to substrate addition via the Echo 555. Samples without inhibitor served as the positive control, whereas samples without enzyme served as the negative control. Plates were read once per minute for 60 min at ambient temperature using a Spectra Max Gemini EM (Molecular Devices, Sunnyvale, CA) at excitation/emission wavelengths of 500/593 nm. In the final optimized assay, manual pipetting steps were replaced with automated liquid handling using a Biomek FX (Beckman Coulter, Brea, CA).
Solubility scanner
A BD Gentest solubility scanner (Becton Dickinson Biosciences, Mississauga, Ontario, Canada) was used to assess compound aggregation state and solubility. Prior to the experiment, the laser was calibrated and the injection loop was washed 96 times with PBS. Ninety-six–well plates (Costar; Corning Life Sciences, Corning, NY) were loaded with two buffer samples (PBS with 5% DMSO) and 10 wells containing a titration of the compound of interest under the same buffer conditions. The instrument was programmed to run the two PBS samples first, with the first one being a wash and the second serving as the background reading. The solubility limit was defined as the concentration at which the events (i.e., number of particles) were threefold above the background reading.
Surface tension determination
Surface tension was measured using a Cahn DCA-332 dynamic contact analyzer (Thermo Fisher Scientific, Nepean, Ontario, Canada) using the Wilhelmy plate technique. The instrument was calibrated according to the manufacturer’s instructions. The sample probe used was a microscope cover slip attached to a copper clip. The probe was cleaned with methanol, air dried, and flamed with a butane torch several times, taking care to keep the slip 3 cm from the flame tip to avoid cracking. The probe was then attached to the machine. Samples were prepared with varying concentrations of substrate in assay buffer (PBS, 5% DMSO) and analyzed within a 50-mL beaker mounted on the instrument sample stage. Prior to measurements, the probe was manually placed ~1 to 5 mm from the sample surface, ensuring the edge of the probe was parallel with the surface of the solution. After each sample measurement, the sample beaker was washed repeatedly with methanol and rinsed with ultrapure water. To ensure that the instrument was functional, a Triton X-100 control was used starting at a concentration of 0.10% v/v. Critical micelle concentration (CMC) values were determined by fitting the decrease in surface tension to a linear model where the x-intercept (analyte concentration) is defined as the CMC.
Data analyses
In all cases, the rate of the enzymatic reaction (in RFU/min) was determined by performing regression analysis on the linear portion of the kinetic curve (0–60 min). The Michaelis-Menten constant (Km) was found by fitting the hyperbolic curve with the following equation: Y = (Vmax * X) / (Km + X), where X is the concentration of substrate (µM), Y is the measurement of activity (RFU/min), and Vmax is the maximum enzyme velocity (RFU/min). The Z′ factor is a measure of the quality of an assay that takes into account both the signal dynamic range and variation of data. 14 The Z′ was determined using the following equation: Z′ = 1 – (3σmax + 3σmin)/(|µmax – µmin|), where σmax and σmin represent the standard deviations of the positive and negative controls and µmax and µmin represent the mean values of the positive and negative controls. IC50 analysis was performed by normalizing the data to % inhibition using the positive and negative controls and applying the four-parameter logistic equation using GraphPad Prism 5.0 (GraphPad Prism Software, San Diego, CA).
Results and Discussion
Resorufin butyrate can serve as a substrate for lipoprotein lipase
Using bLPL as a model enzyme for triglyceride lipases, we assessed whether resorufin butyrate could be adopted as a soluble and monomeric substrate. An initial experiment in 384-well plates using 12 nM of bLPL enzyme and 2.3 µM substrate in PBS, pH 7.5, 5% DMSO gave a linear increase in fluorescence over 60 min. Control samples without enzyme gave small increases in fluorescence ascribable to background hydrolysis of the substrate ( Fig. 1B ). The activity of the enzyme did not differ between 1% and 10% DMSO (not shown), and thus we elected to use a concentration of 5% to potentiate the monomeric form of the substrate while avoiding atypical concentrations of DMSO. Under these conditions, we examined the reaction velocity at various concentrations of substrate to calculate the Km value ( Fig. 1C ). However, since full saturation was not reached due to insolubility of the substrate at the highest concentration (187.5 µM), the calculated value of ~40 µM should be considered only an estimate.
To maximize the probability of maintaining substrate in a soluble and monomeric form, we sought to determine the minimal amount of resorufin butyrate the assay could tolerate. Thus, we assessed assay quality at 10 nM of bLPL and a range of substrate concentrations. Concentrations below 111 nM of substrate gave coefficient of variation (CV) values >15 and thus were not considered for further development. At concentrations of 111 nM and above, acceptable CV values were obtained and the signal to background (S/B) was >8 at the three concentrations tested ( Fig. 1D ). Although increased amounts of enzyme gave more signal (not shown), we wished to minimize enzyme concentration to maximize the assay’s dynamic range. However, further dilutions of enzyme yielded a highly variable assay and were thus not considered further (not shown). For the final assay conditions, we selected 10 nM of bLPL and 100 nM of resorufin butyrate to maximize assay sensitivity and substrate behavior.
Resorufin butyrate is soluble and monomeric under assay conditions
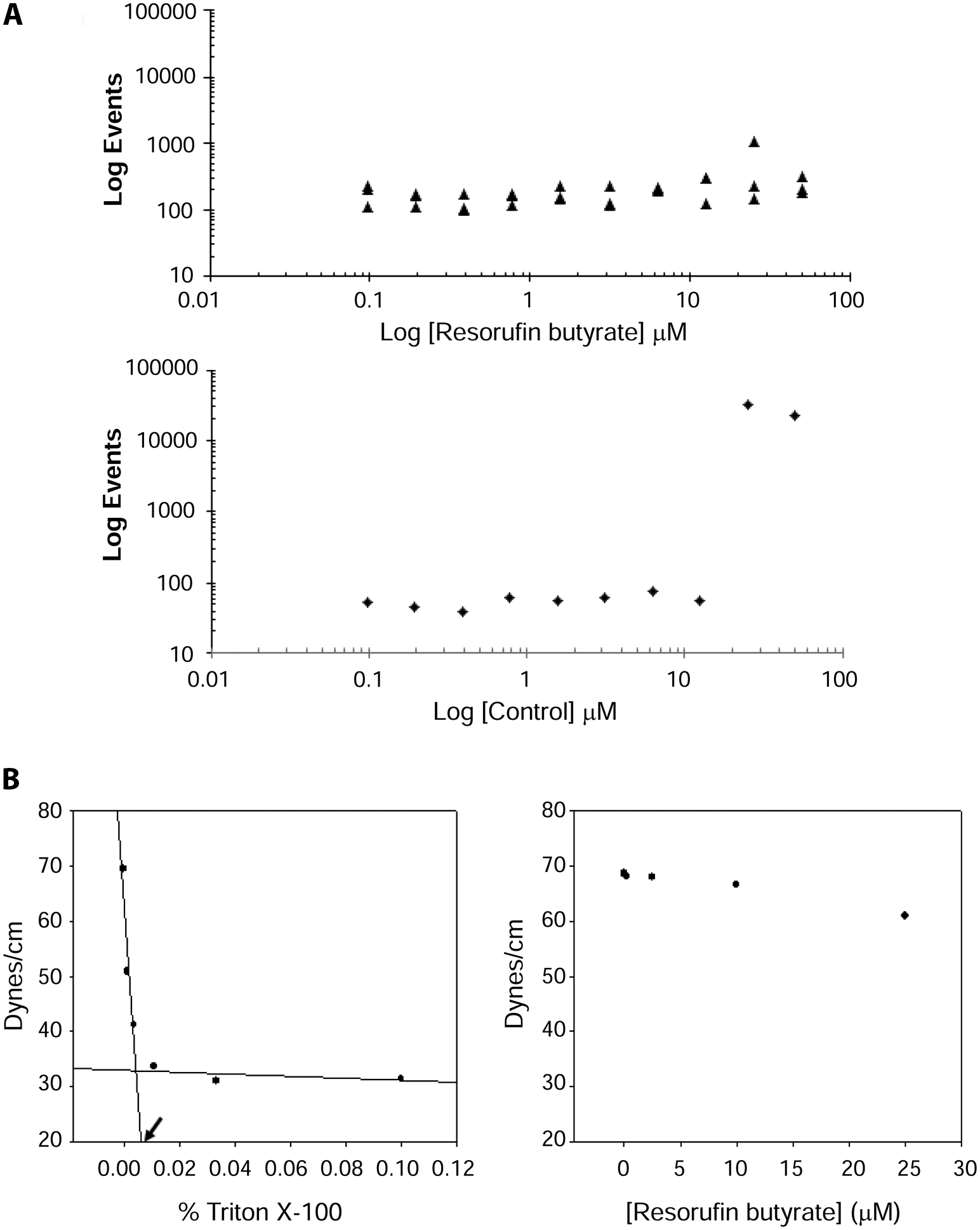
Having established assay conditions, we sought to determine whether resorufin butyrate was monomeric at the selected concentration (100 nM) using static light scattering as a first approach. Under assay buffer conditions, no particles were observed up to a concentration of 50 µM, suggesting that the molecule is soluble at all concentrations tested ( Fig. 2A , upper panel). In contrast, particles were detected for our in-house control compound at concentrations >12.5 µM, in agreement with historical results ( Fig. 2A , lower panel). Although encouraging, the smallest diameter detectable with the instrument used (Gentest solubility scanner, BD) is ~0.2 micrometers, and thus the presence of soluble resorufin butyrate aggregates (i.e., micelles) with smaller diameters is not excluded.
Resorufin butyrate is soluble and monomeric under assay conditions. (
To confirm that resorufin butyrate was not forming micelles under assay conditions, titrations of this molecule were followed by surface tension measurements, the gold-standard method for determining a molecule’s CMC. The profile for Triton X-100, a micelle-forming detergent, is repeated here as an assay standard ( Fig. 2B , left panel). Increasing concentrations of Triton X-100 resulted in a progressive decrease in surface tension until the CMC was approached, at which point the changes in surface tension leveled off. In agreement with the literature (e.g., Zhang et al. 15 ), we report a CMC of Triton X-100 of 0.006% (v/v). When resorufin butyrate was titrated from 25 µM in assay buffer, no substantial change in surface tension was noted as it remained approximately equal to that of assay buffer ( Fig. 2B , right panel), suggesting that this molecule does not form micelles. Importantly, the optimized assay uses 100 nM of substrate, making it very unlikely for particles to form at this concentration. Together, the combined data indicate that resorufin butyrate is soluble and monomeric under assay conditions and can be employed in a particle-free lipase assay. This format is a key distinguishing feature of this assay since particle-based substrates can introduce artifacts that can confound data interpretation.9,10
Resorufin butyrate can be adapted as a robust screening assay
Having established that resorufin butyrate can be used as a substrate for bLPL and that the molecule can indeed be employed as a particle-free substrate, we sought to determine whether the assay was robust enough for inhibitor screening. Using a 384-well format, a whole-plate experiment was run to assess signal variability. Specifically, all wells contained the negative control (enzyme without inhibitor) except for half of the outside wells, which were reserved for the positive control (sample without enzyme). The assay showed reproducibility within each control and yielded an S/B = 10.5, a CV = 4.5%, and a Z′ = 0.82, indicating an assay suitable for single-point screening (data not shown).
To further assess the robustness of the assay, we used a proprietary Merck lipase inhibitor control to generate IC50 curves on three separate days (n = 3). M-352, a sulfonyl fluoride–containing compound, was incubated with enzyme for 30 min prior to substrate addition. The IC50 value generated was 301 ± 39 nM ( Fig. 3 ), which is within twofold of in-house historical values obtained for unpurified human LPL in particle-based assays ( Table 1 ). To test assay reproducibility, IC50 values for five propitiatory LPL inhibitors (representing different structural classes), including M-352, were generated on two separate days. Importantly, the IC50 values had interday shifts of less than threefold, and enzyme and substrate appeared to be stable over time. Furthermore, the IC50 values generated with the particle-free assay were similar to those obtained using our in-house particle-based assay (all potencies were within threefold; Table 1 ). The good correlation between the two assays also suggested that the signal seen in the resorufin assay is specific to bLPL rather than potentially contaminating lipases.
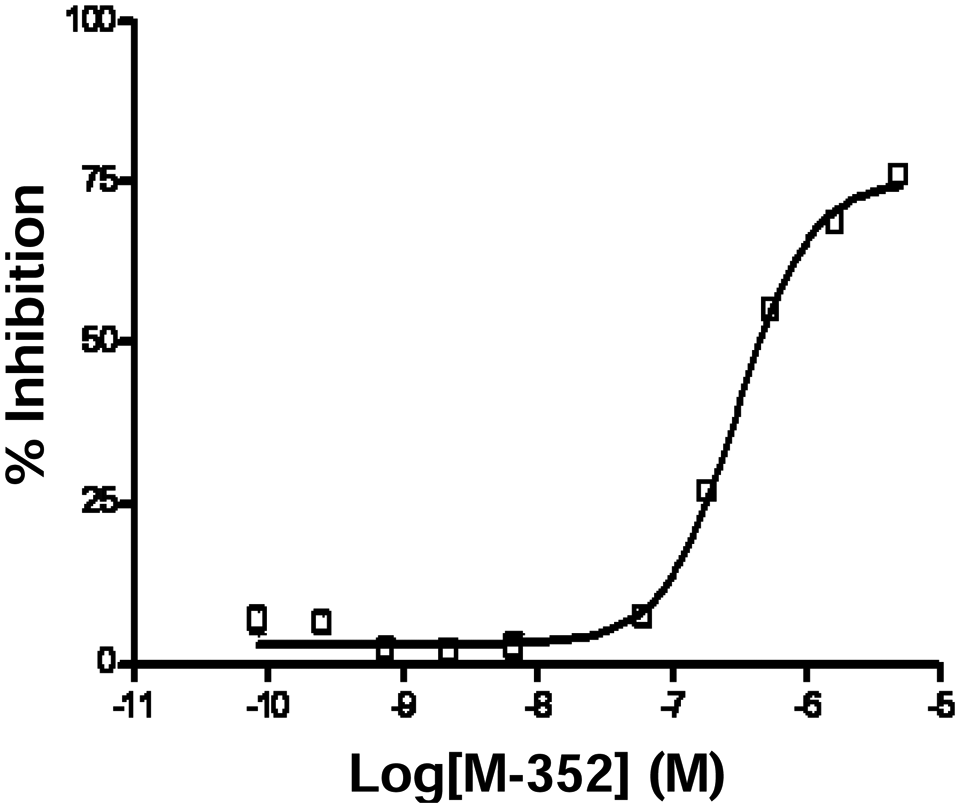
Resorufin butyrate can be adapted as a robust screening assay. Pipetting steps were automated on a Biomek FX liquid handler under the optimized conditions of phosphate-buffered saline (PBS), pH 7.5, 5% DMSO, with resorufin butyrate (100 nM) and bovine lipoprotein lipase (bLPL; 10 nM) in 384-well format. The assay is suitable for the generation of IC50 values where the values obtained had minimal variation between days. The error bars represent the mean ± SD, and the curves were fit using the four-parameter logistics equation.
IC50 Values of Merck Inhibitors with Two Different Assays
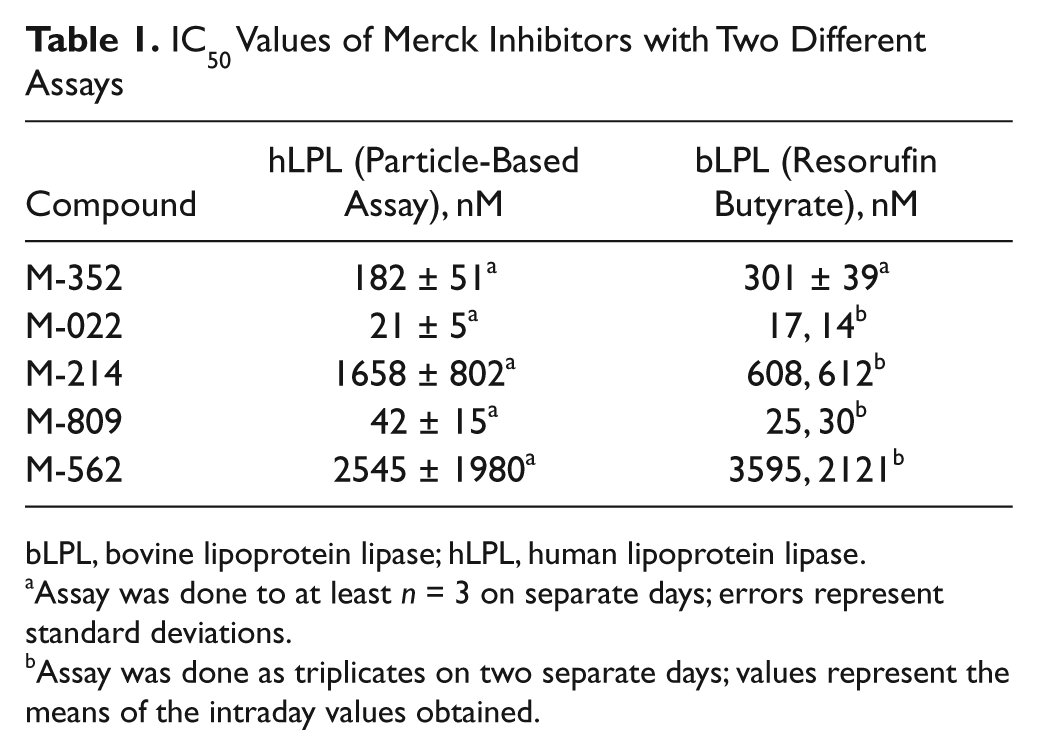
bLPL, bovine lipoprotein lipase; hLPL, human lipoprotein lipase.
Assay was done to at least n = 3 on separate days; errors represent standard deviations.
Assay was done as triplicates on two separate days; values represent the means of the intraday values obtained.
Here, we have expanded the use of resorufin butyrate as a substrate for lipases in three important ways. First, by demonstrating that resorufin butyrate can be processed by bLPL, we extend its use into an important therapeutic group of enzymes, the triglyceride lipase family. Second, we define conditions where the substrate is soluble and monomeric. This outcome was critical to our goals and enabled a particle-free lipase assay, allowing one to profile compounds in a manner that avoids the artifacts associated with lipid-particle substrates. Finally, we have developed and validated a high-throughput assay that is robust, simple, and inexpensive. In fact, in terms of substrate, the cost per 50-µL sample is <0.001 cents, which would translate to a cost of $10 for a high-throughput screening campaign consisting of 1 million wells. Indeed, the assay is well suited to high-throughput efforts given its robust characteristics (S/B > 10, CV < 5%, and a Z′ > 0.8), stability of reagents, and acceptable interday variability (potencies varied less than threefold). Finally, although the current assay was developed in 384 wells, the screen is likely scalable to 1536- or 3456-well plate densities.
We envision applying the current assay as either a simple and inexpensive primary screen to identify novel enzyme modulators or as a follow-up strategy to eliminate false positives from hit lists derived from lipid-based substrate screens. In either mode, the assay should provide a useful tool for the discovery of therapeutic molecules.