
Abstract
The authors have used a surface plasmon resonance (SPR)–based biosensor approach to identify and characterize compounds with a unique binding mode to protein kinases. Biacore was used to characterize hits from an enzymatic high-throughput screen of the Tec family tyrosine kinase, IL2-inducible T cell kinase (ITK). Complex binding kinetics was observed for some compounds, which led to identification of compounds that bound simultaneously at both the adenosine triphosphate (ATP) binding site and a second, allosteric site on ITK. The presence of the second binding site was confirmed by X-ray crystallography. The second site is located in the N-terminal lobe of the protein kinase catalytic domain, adjacent to but distinct from the ATP site. To enable rapid optimization of binding properties, a competition-based Biacore assay has been developed to successfully identify second site noncompetitive binders that have been confirmed by X-ray crystallographic studies. The authors have found that SPR technology is a key method for rapid identification of compounds with dual-site modes of action.
Introduction
Protein kinases belong among the most intensively studied classes of enzymes. The vast majority of protein kinase inhibitors bind at the adenosine triphosphate (ATP) binding site. The therapeutic potential of protein kinase inhibitors can be limited due to the inherent difficulties in developing selective kinase inhibitors. Similarities between protein kinases in protein sequence and structure at the ATP binding site often compromise inhibitor selectivity, which is important for minimizing interference with other biochemical pathways. 1 As there are more than 500 protein kinases in the human genome, targeting the relatively conserved ATP binding pocket creates significant difficulties in designing selective kinase inhibitors. 2
Although the vast majority of kinase inhibitors target the ATP binding site, they need to compete with high levels of ATP in the cell to be effective. In contrast, allosteric modulators of protein kinases act at binding sites distinct from the ATP site3-6 and thus display different selectivity profiles over conventional ATP site binders. Allosteric kinase ligands can also increase the range of functions with both allosteric kinase activators and inhibitors being reported. 7 Moreover, non-ATP competitive ligands, binding at sites distinct from the ATP binding site, may theoretically have better therapeutic efficiencies as they are not competing with the high levels of ATP in the cellular environment. 8 Technologies that can systematically and cost-effectively identify allosteric kinase modulators may open up new therapeutic opportunities for protein kinases.
Conventional high-throughput methods for screening protein kinases usually employ biochemical and whole-cell assays biased against inhibition of ATP binding by compounds. We have developed surface plasmon resonance (SPR) assays as an alternative screening method to accelerate the kinase drug discovery process. Over the past two decades, SPR-based biosensors have become a powerful tool for the characterization of biomolecular interactions.9-12 Recent developments have enabled SPR to be used for the determination of real-time interaction kinetics for molecules as small as 100 Da binding to a protein immobilized on a chip. 13 In this study, SPR-based biosensor Biacore (GE Healthcare, Uppsala, Sweden) technology was used to discover and characterize compounds with a unique binding mode to the human cytoplasmic Tec family tyrosine kinase, IL2-inducible T cell kinase (ITK). ITK plays an important role in the production of cytokines such as interleukin (IL)–2, IL-4, IL-5, IL-10, and IL-13. 14 Therefore, due to the role of ITK in T cell signaling, inhibition of the kinase could be the basis of treatment of inflammation disorders such as allergic asthma and psoriasis.15,16
Materials and Methods
Materials
The Biacore 3000 and S51 optical biosensors, CM5 sensor chips, and the amine-coupling kit were obtained from GE Healthcare. The human tyrosine kinase was prepared in house (described in the next subsection). Immunopure streptavidin was purchased from Pierce (Rockford, IL). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Cloning, Expression, and Purification of Human Tyrosine Kinase, ITK
The kinase domain of human tyrosine kinase, ITK, was cloned into pFastbac1 (Invitrogen, Carlsbad, CA) with an N-terminal BAP (biotin acceptor protein)17,18 tag and a C-terminal His6 tag. Within the pFastbac vector, the BirA gene was also inserted downstream of a p10 promoter. Recombinant baculovirus was generated using the Bac-to-Bac baculovirus expression system (Invitrogen). Protein was expressed by infection of recombinant baculovirus with T.ni High5 insect cells in 1-L Erlenmeyer flasks using Excel 405 media (SAFC Biosciences, Lenexa, KS) with a harvest time of 72 h. Coexpression with BirA is essential in obtaining biotinylation of the N-terminal BAP tag. Human tyrosine kinase was purified using nickel affinity chromatography. The hexahistidine tag was then removed by overnight incubation at 4 °C with 15 U/mg bovine thrombin (Sigma-Aldrich). Subsequent purification by size exclusion chromatography on a Superdex 200 (16/60) column (GE Healthcare) yielded a homogeneous preparation.
Biochemical Assay
Enzymatic activity of ITK was determined using the ADP-Glo Kinase Assay 19 (Promega, Madison, WI) and reagents provided with Promega’s Kinase Glo Plus Kit following protocols recommended by the manufacturer. Recombinant human ITK was purchased from Invitrogen. The reactions were performed in the assay buffer (50 mM HEPES [pH 7.5], 5 mM MgCl2, 0.1% F-127, 1 mM dithiothreitol [DTT]) in a 384-well plate format. The reactions were carried out at room temperature in a total volume of 40 µL for 10 min with 100 µM ATP and at saturated substrate concentrations. Enzyme and substrate solutions were incubated for 2 h before Kinase Glo Plus reagents were added and incubated for 120 min. The plate was read by a Generation IV LEADseeker (Amersham Biosciences, UK Ltd.) with 4-s image time (coincident averaging).
Crystallization and X-Ray Crystallography
Crystals of ITK were grown by the hanging drop vapor diffusion technique. ITK protein at a concentration of ≈10 mg/mL was preincubated with 5 mM inhibitor prior to co-crystallization trials. Optimal conditions for crystal growth were 100 mM Mg acetate, 0.1 M HEPES at pH 7.0, and 10% PEG 3350 as the precipitating agent at 4 °C. The crystals were flash frozen after transfer into a cryo-protectant solution containing mother liquor plus 10% ethylene glycol prior to data collection on beamline ID23eh1 at the European Synchrotron Radiation Facility (ESRF). Crystals typically diffracted between 1.4 and 2.0 Å resolution and belonged to space group P21 with unit cell dimensions a = 40.9 Å, b = 68.3 Å, c = 49.4 Å, β = 107°. Data were processed and scaled using the program MOSFLM within the CCP4 20 suite of programs. Structure solution by molecular replacement techniques and refinement of the complexes using the REFMAC program yielded structures with crystallographic R factors <23% with very good geometry. Full details of the structural aspects of this work will be published at a later date.
Protein Immobilization
Streptavidin was immobilized on a CM5 chip at 25 °C using standard amine coupling chemistry at a flow rate of 10 µL/min. The dextran surface was activated by a 12-min injection of a 1:1 ratio of 0.4 M EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride) and 0.1 M NHS (N-hydroxysuccinimide) followed by a 15-min injection of streptavidin in 10 mM sodium acetate, pH 4.5. All unreacted groups on the surface were blocked by a 7-min injection of 1 M ethanolamine, pH 8.5. HBS (10 mM HEPES, 150 mM NaCl, pH 7.4) was used as a running buffer for immobilization. ITK was captured via a biotin tag in a kinase buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM DTT, 5 mM MgCl2, 1 mM MnCl2, 5% DMSO, and 0.005% Tween 20) at a flow rate of 30 µL/min. The highest density of captured protein obtained was approximately ~9000 response units (RU). However, to minimize mass transport effects, the amount of captured protein was reduced to 1000 to 3000 RU depending on the molecular weight of the compound and the kinetic profile of binding.
Kinetic Characterization
Unless stated otherwise, all binding experiments were carried out using the S51 instrument at 4 °C to increase protein stability over time. Compounds were solubilized in 100% DMSO in a stock concentration of up to 50 mM and then diluted further in DMSO and running buffer to obtain the required concentration necessary for a good data quality specifically adjusted around the KD value for each compound. 21 Compounds were injected at a flow rate of 90 µL/min, and association was measured for 1 min and dissociation for 2 to 15 min depending on the off-rate of each compound. Competition experiments were carried out in the presence of compound A at a 200-nM concentration diluted in the running buffer.
Temperature-Dependent Kinetic Characterization
Experiments were performed under the same conditions as described in the “Kinetic Characterization” section using a Biacore 3000 instrument. Interaction was measured for binding of compounds E and F in threefold dilution series at concentrations of 13.33 to 0.05 µM and 3.33 to 0.014 µM, respectively, at a flow rate of 50 µL/min. Association was measured for 1 min and dissociation for 3 min. Experiments were carried out at four temperatures—6, 12, 18, and 24 °C—to determine whether the affinity of compound binding was temperature dependent.
SPR Data Analysis
Prior to kinetic analysis, all data were processed and double referenced for a blank streptavidin reference surface and blank injections of running buffer to minimize the influence of baseline drift upon the binding kinetics. For analysis, we used Scrubber software (version 2.0; BioLogic Software, Campbell, Australia). For easier data comparison, all sensorgrams were normalized for protein capture level 3000 RU and Mw of ligands 200 Da.
Results
Assay Optimization
To achieve good-quality SPR data for any biomolecular interaction, the target protein has to be immobilized in a way that maintains its activity, and reaction conditions should facilitate as closely as possible the native conformation of the target. Therefore, a biotinylated tag was engineered on the N-terminus of the ITK catalytic domain to ensure an oriented capture to a biosensor chip via immobilized streptavidin. This capture method provides an efficient protein attachment at neutral pH compared to traditional amine-coupling methods, where the protein is subjected to an acidic environment, which in turn can deactivate the protein. 22 Alternative immobilization methods using protein modification such as minimal biotinylation, where biotin can chemically react with proteins 21 under neutral pH, do not guarantee a specific orientation of the protein on the chip and may also inhibit the kinase binding site if these contain reactive amine/carboxyl groups.
For the experiments, we used DMSO as a solvent for compound solubilization. To increase solubility of compounds in the running buffer, the DMSO concentration was kept at 5%. Because the concentration of DMSO exceeded the typical levels used for biochemical assays (0.1%), it was necessary to assess that this concentration did not interfere with binding properties of the target or influence its activity. 23 Therefore, we measured binding of ATP to the kinase in running buffer containing various DMSO concentrations: 1%, 3%, and 5%. Figure 1A shows equilibrium fits of ATP binding under each DMSO concentration. The increasing DMSO concentrations increased affinity of the kinase to ATP from 26.7 µM to 16.3 µM. As there was no significant negative effect of high solvent concentration observed, further experiments were performed at 5% DMSO.
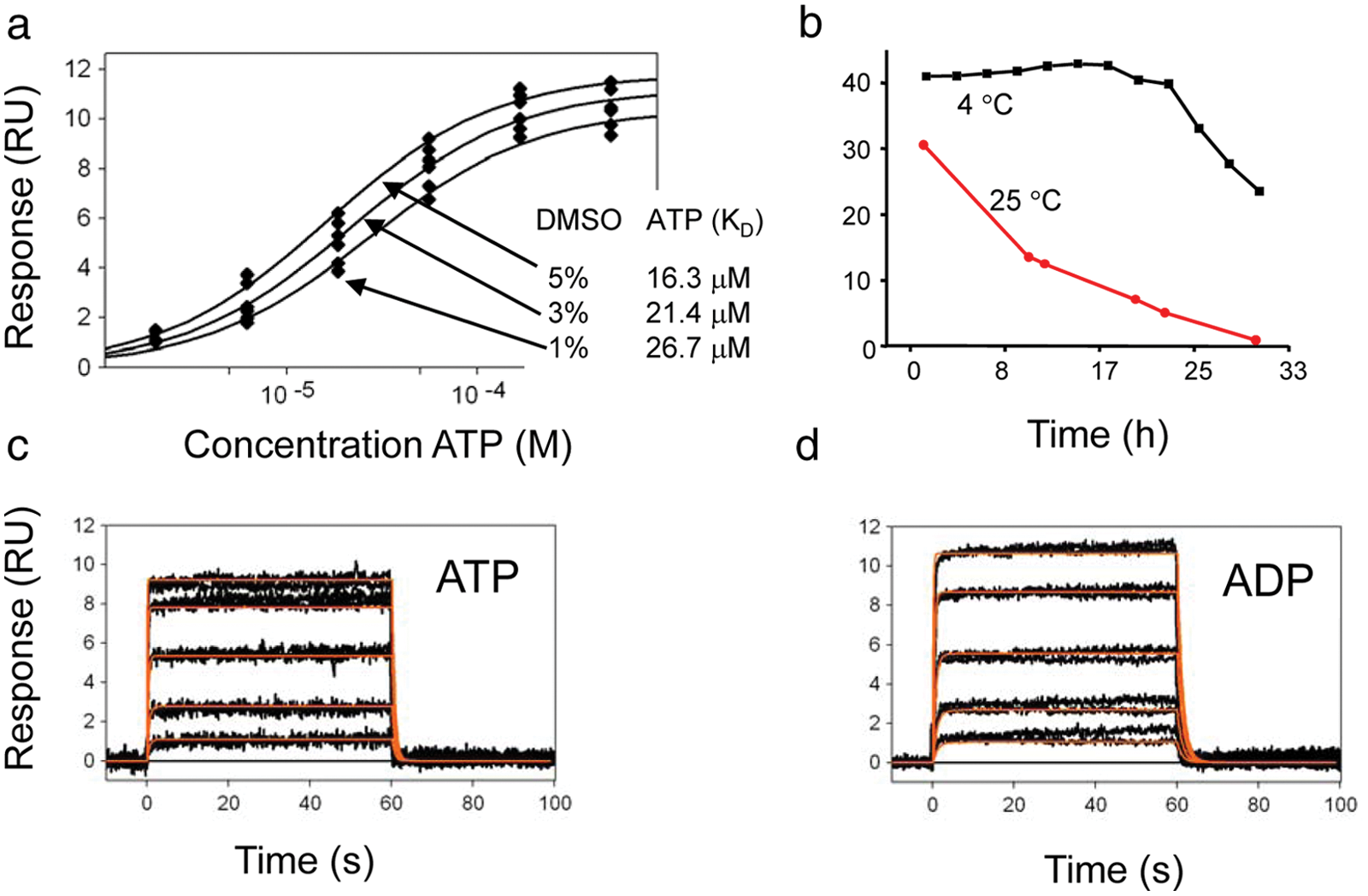
(A) DMSO effect screen: equilibrium fit of adenosine triphosphate (ATP) binding responses (ATP injected at 0.4, 1.23, 3.7, 11.1, 33.3, and 100 µM). (B) Stability test: ATP injected at 100 µM over the IL2-inducible T cell kinase (ITK) surface at 25 °C (red) and 4 °C (black). (C) ATP binding responses (black) with kinetic fit (red; concentrations: 2, 6.17, 18.5, 55.5, and 166 µM). (D) Adenosine diphosphate (ADP) binding responses (black) with kinetic fit (red; concentrations: 0.4, 1.23, 3.7, 11.1, and 33.3 µM). RU, response units.
The stability of the protein immobilized on the chip is necessary for experiments where a large number of compounds are screened. In this case, we tested whether the kinase maintains its activity under room temperature conditions. We repeatedly injected ATP at a 100-µM concentration over the surface with immobilized protein for 32 h. As shown in Figure 1B , the binding response to ITK rapidly decreased when the binding was measured at 25 °C, and by the end of the experiment, no ATP response was observed. By lowering the temperature to 4 °C, the protein retained ATP binding activity, so at the end of a 32-h run, we observed only a 50% decrease in ATP binding response. All further experiments were conducted at 4 °C.
The functionality of the SPR assay was confirmed by measuring binding of nucleotides ATP and adenosine diphosphate (ADP) with affinities 16.4 µM and 4.3 µM, respectively ( Fig. 1C , D ), comparable with affinities measured by biochemical assays (data not shown). The kinetics was fitted to a 1:1 binding model, yielding kinetic constants ka = 9.1 (±0.4) × 104 M−1 s−1 and kd = 1.5 (±0.4) s−1 for ATP and ka = 1.8 (±0.2) × 104 M−1 s−1 and kd = 0.8 (±0.1) s–1 for ADP. The functionality was further tested with a known high-affinity inhibitor, BMS-5096744 (compound A) 24 ( Fig. 2 ). The kinetics for this compound was fitted to a 1:1 binding model, 25 including mass transport limitation, giving an association rate constant ka = 2.8 (±0.4) × 105 M−1 s−1 and dissociation rate constant kd = 9.7 (±0.5) × 10−5 s−1. Because it was not possible to regenerate the interaction between compound A and ITK, compound A was injected from the lowest (1.2 nM) to the highest (370 nM) concentrations using a threefold dilution series, and when fitted, the data were normalized for Rmax of each concentration. 26
Competition Assay
Initially, we used the SPR assay to confirm hits identified in a high-throughput biochemical assay bound to ITK, and once this was confirmed, we kinetically characterized the confirmed hits. Most of the binders showed a typical 1:1 interaction and stoichiometry, which was possible to fit to a 1:1 binding model, as shown with some examples (compounds A–C) in Figure 2 . The kinetic constants for one-site binders are summarized in Table 1 . Complexity in the dissociation phase and increasing stoichiometry value were apparent from binding sensorgrams shown for compound D ( Fig. 2 ). Crystallization of this compound with ITK revealed that this compound binds to two distinct sites on the kinase. We fitted the sensorgrams to a two-site binding model to determine binding kinetics and affinities for each site separately. To determine kinetics of binding to the second site only, we measured binding of compound D in the presence of a high-affinity ATP site inhibitor. Compound A fully occupied the ATP site of ITK, with high affinity and slow off-rate, effectively blocking this site to other ligands, and therefore all competition studies were conducted with compound A. Therefore, any observed binding of compounds can be explained by them only interacting with alternative binding sites. Using this competition assay, simple binding kinetics was observed for compound D with kinetic values summarized in Table 1 . Compounds E, F, and G were then selected based on similar chemical structures to compound D (data not shown), and binding kinetics and affinity were calculated using the same method, where in the absence of compound A, binding was fitted to a two-site model and in the presence of competitor compound fitted to a 1:1 model to determine kinetics of binding to the second site. All compounds except compound F have been selected for further crystallographic studies, which confirmed single- or multiple-site binding and the discovery of a novel binding site on ITK ( Fig. 2 ).
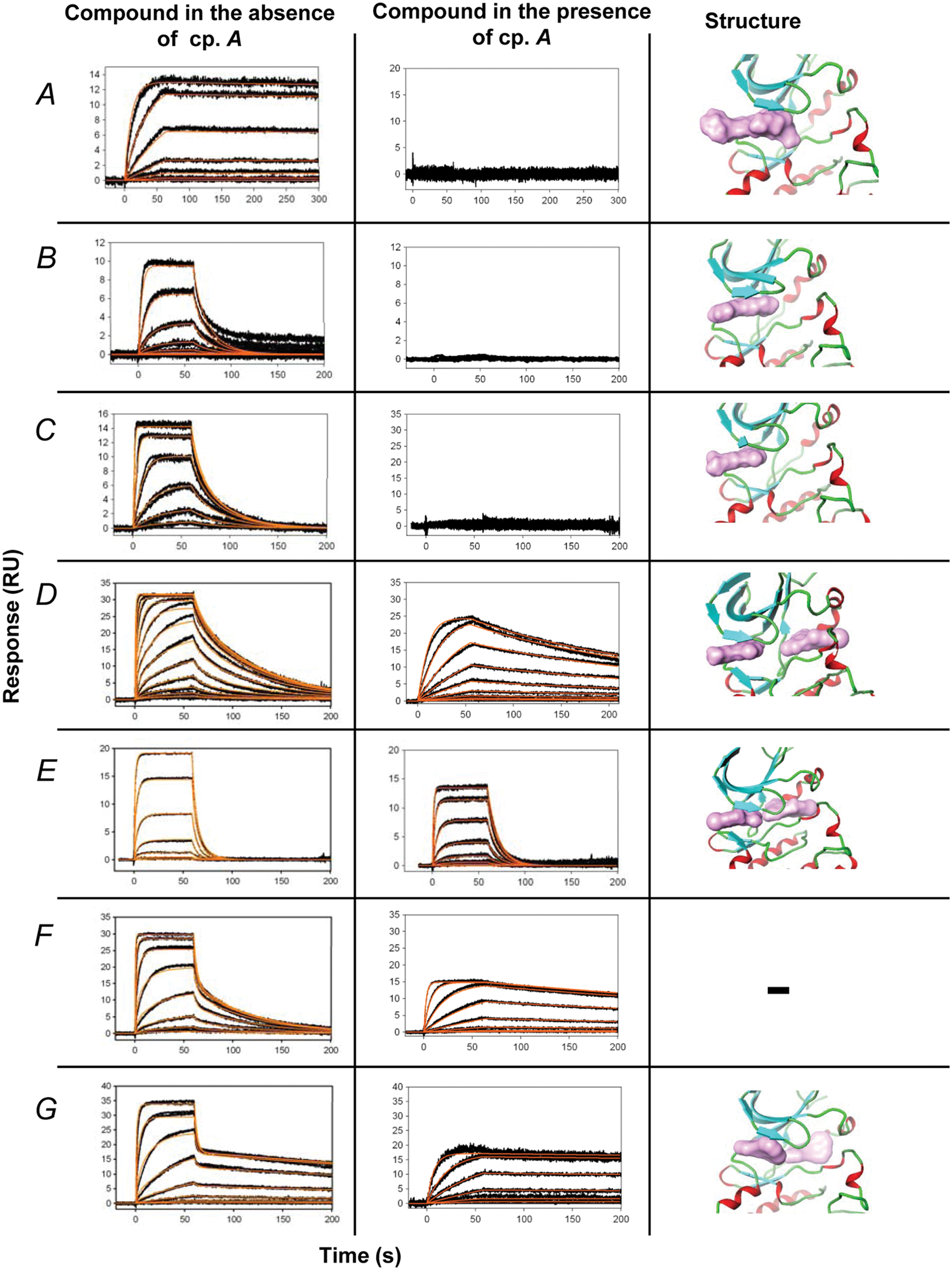
Compounds binding to the immobilized IL2-inducible T cell kinase (ITK). Black curve represents the measured responses, red curve the kinetic fit. Compounds were injected in duplicates in concentration series in the presence and absence of 200 nM inhibitor (compound A). Next to each binding trace is a figure of the X-ray crystallographic complex structure of the corresponding compound bound to ITK for the compounds for which X-ray crystallographic data were obtained. The surface area of each compound is represented in mauve. The structure of ITK is represented as alpha helices (red), beta sheets (turquoise), and loops (green). Compounds were injected at threefold concentration dilution series (compound D, twofold). The top concentrations were as follows: compound A, 370 nM; compounds B and C, 1 µM; compound D, 5.7 µM; compound E, 13.3 µM; compound F, 10 µM; compound G, 5 µM. RU, response units.
Kinetic, Affinity, and IC50 Parameters with Standard Deviation of Kinetic Fits (in Parentheses) Measured for Compounds A to G Binding to IL2-Inducible T Cell Kinase (ITK)
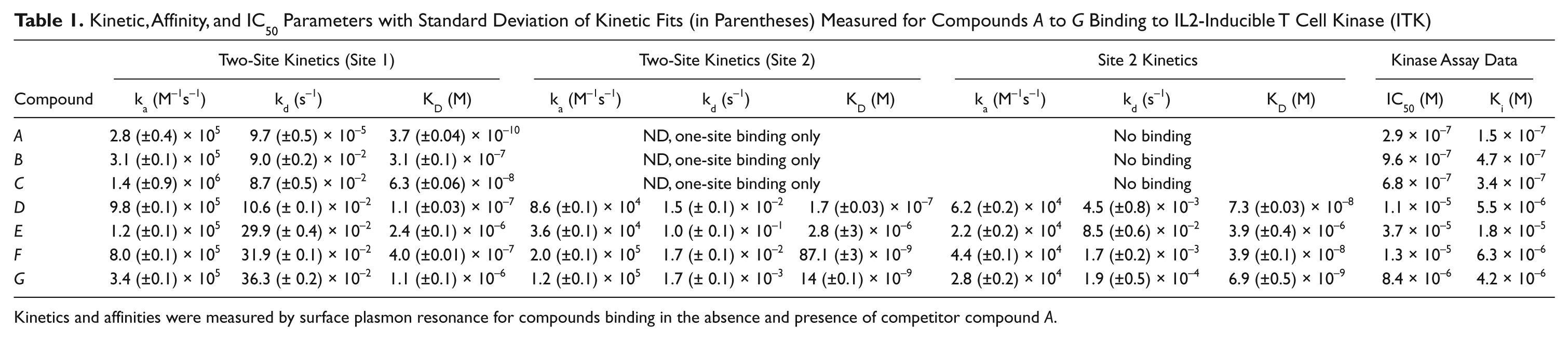
Kinetics and affinities were measured by surface plasmon resonance for compounds binding in the absence and presence of competitor compound A.
Kinetic values are summarized in Table 1 together with IC50 values determined by biochemical Kinase Glo enzyme assay (Promega). The higher affinity values measured by the SPR assay are probably the result of the low temperature used to measure binding, due to instability of ITK at room temperature for a longer period. However, values for affinities measured to two-site binders do not correlate with enzyme assays. This is probably explained by the mechanism of ATP displacement on which the biochemical assay is based and does not necessarily detect binding to the second site on the kinase.
Temperature Dependence of Binding Kinetics
As mentioned above, to increase protein stability, all SPR experiments were carried out at 4 °C. It has been shown previously27,28 that the kinetics and affinity of interactions can be influenced by temperature. Therefore, we selected two compounds (E and F) and measured their binding to the kinase in the presence and absence of compound A at four different temperatures: 6, 12, 18, and 24 °C. Figure 3 shows binding sensorgrams for both compounds at these temperatures. Compound E exhibits a 1:1 interaction profile both in the presence or absence of competitor compound ( Fig. 3A , B ) at all temperatures, indicating that compound E has similar affinities at both sites. Compound F shows complex behavior at low temperatures in the absence of compound A ( Fig. 3C , 6 °C and 12 °C), but at increasing temperatures, the complexity of binding disappears as the binding affinities for both sites become weaker. For binding of compounds in the absence of compound A, the sensorgrams were fitted to a two-site model. However, with increasing temperature, especially for compound E, the model failed to recognize two binding sites and became unreliable. This is mainly caused by rapid change of kinetics and affinity toward higher temperatures and also partial instability of the kinase at higher temperatures ( Fig. 1 ). In contrast, the data collected for binding in the presence of competitor compound A show very clear 1:1 binding kinetics and different on/off-rates between low and high temperatures, as the kinase is stabilized by the presence of compound A. Compound F also shows a significant difference in the dissociation rate at lower temperatures, where the measured kd in the presence of compound A is ~20 times slower (3.1 × 10−3 s−1) compared to 24 °C (6.7 × 10−2 s−1) ( Table 2 ).
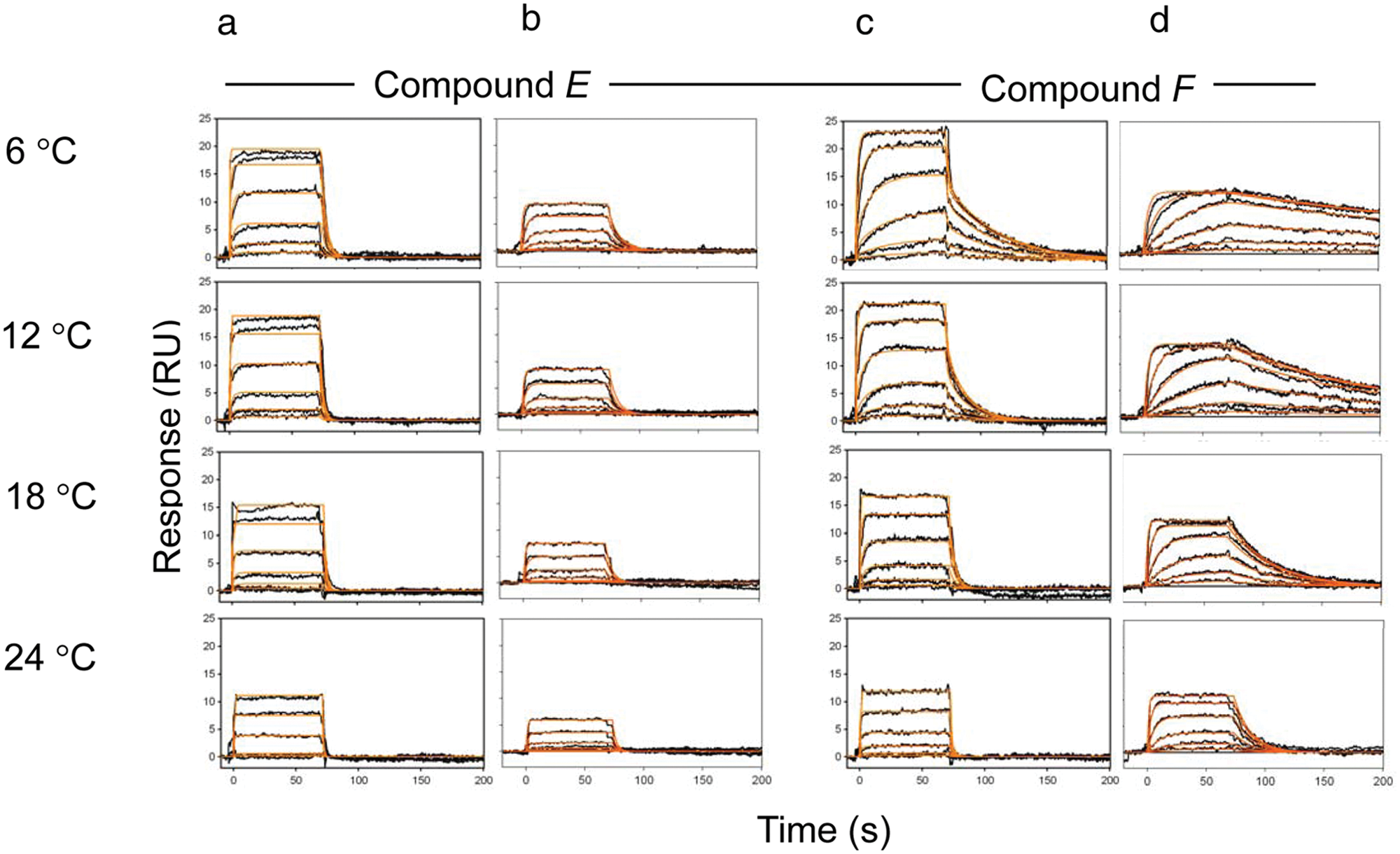
Binding sensorgrams measured for compounds binding to IL2-inducible T cell kinase (ITK), at four temperatures (6, 12, 18, and 24 °C). (A) Compound E in the absence of an adenosine triphosphate (ATP) site inhibitor. (B) Compound E in the presence of an ATP site inhibitor, including kinetic fits (red). Top concentration injected—13.3 µM in a threefold dilution series. (C) Compound F in the absence of the ATP site inhibitor. (D) Compound F in the presence of the ATP site inhibitor, including kinetic fits (red). Top concentration injected—3.3 µM in threefold dilution series. RU, response units.
Kinetic and Affinity Constants with Standard Deviation Of Kinetic Fits (in Parentheses) for Compounds E and F Binding to IL2-Inducible T Cell Kinase (ITK) at Four Temperatures: 6 to 24 °C
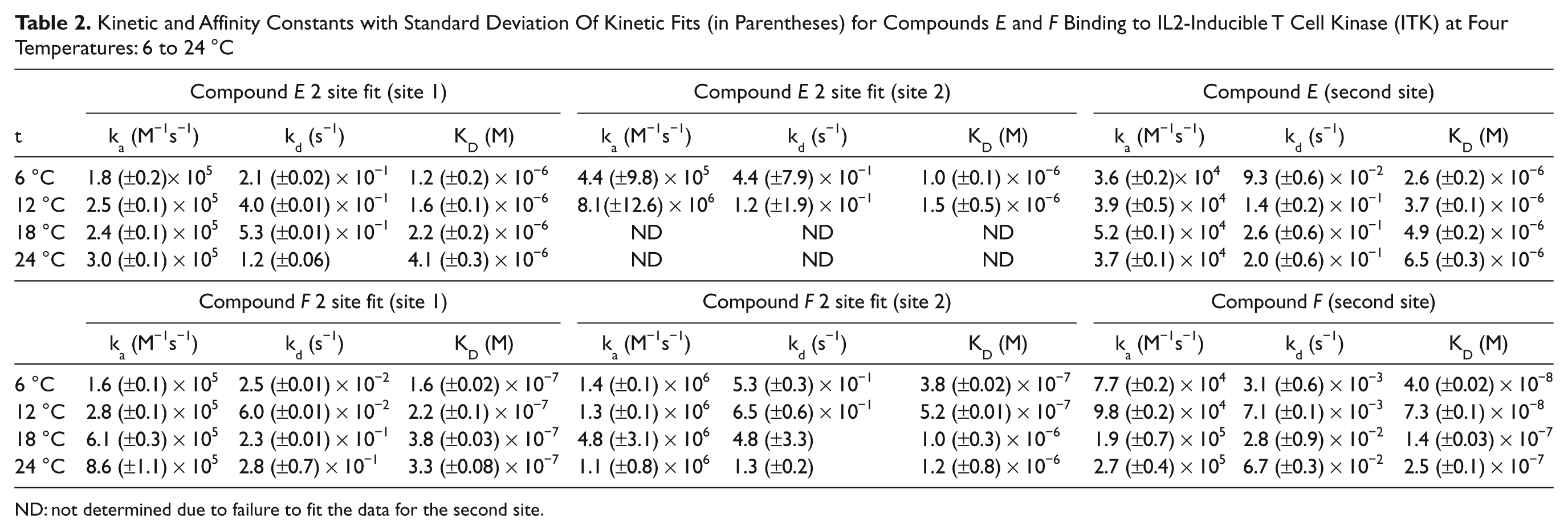
ND: not determined due to failure to fit the data for the second site.
Increased stoichiometry of binding for both compounds in the absence of compound A is observed at lower temperatures (6 °C and 12 °C). Binding stoichiometry for compound E at low temperatures suggests more than one interaction site. The saturating response at the highest concentration (23 RU) is ~2 times higher compared to theoretical Rmax based on the level of immobilized protein and ATP binding (12 RU).
29
However, as temperature increases, the stoichiometry of the binding interaction gets closer to 1:1. We consider this observation to be an artifact of protein instability, as previously observed at 25 °C (
Discussion
In this study, we used SPR-based biosensor technology to identify and characterize compounds with a unique, dual-binding mode to the nonreceptor tyrosine kinase, ITK. Complex binding kinetics was observed for some compounds, which led to the identification of compounds interacting not only with the ATP binding site but also with a novel second site.
It is important to note that SPR technology is based on the change of optical properties near the sensor surface caused by any mass change, 30 and therefore complex binding kinetics is difficult to interpret because in most cases it is an artifact of suboptimal assay design. 21 Several cases where the complexity of binding led to the identification of a novel binding site are described in this study. However, before we decide if the complex binding is due to the molecular mechanism of interaction, the quality of data and assay design has to be carefully optimized.
The activity of an immobilized target protein has to be maintained at a high level. This can be achieved by selecting suitable immobilization technique and assay conditions. Generally, to measure binding of small-molecule inhibitors, these compounds have to be solubilized in a suitable solvent as their solubility in aqueous buffers is often low. Presence of any artificial solvent may be critical for protein activity and therefore should be identified. Long-term stability of the protein is also important, and therefore including control compound injections within the experiment is crucial to assess protein stability.
Compounds binding to two sites on the kinase showed increased stoichiometry compared to compounds binding at the ATP site alone. Complexity of binding was related to the binding affinities at each site, and therefore compounds with similar affinities at both ATP and the second site could be mistaken for one-site binders. As the binding stoichiometry could be the main parameter to decide whether the compound is binding to one or more sites, depending on the protein activity, this information may be misleading. In our study, we found strong dependence of protein activity and kinetics of compound binding on temperature. The apparent slow off-rate measured for compound F at 4 °C significantly increased with elevated temperature, resulting in a ~6-fold decrease in affinity, whereas compound E showed decreased binding affinity of only 2-fold. Binding stoichiometry was significantly different for compounds binding in the absence or presence of an ATP site inhibitor when the interaction was measured at low temperature. At higher temperatures, protein activity decreased, and the stoichiometry was similar to binding restricted to just one site. These data suggest that long-term protein stability is essential for accurate stoichiometry measurements when analyzing a large number of analytes on the same target. Increased stoichiometry and complex binding can both provide evidence for multisite binders, but as both these parameters can be influenced by other factors (e.g., experimental design, temperature, protein activity), the only way to unequivocally determine multisite binding is by a competition assay.
We confirmed that the SPR technology is a suitable method for determination of multisite binders by selecting six compounds binding either to one or both sites on the kinase for which we obtained X-ray crystallography structures. In all cases, we found excellent agreement between X-ray crystallography and SPR in the identification of dual-site binders. This observation strengthened our confidence to use the SPR competition assay to identify second site binders. Using the SPR assay, we were able to identify compounds that bound to the kinase and also provide kinetic and affinity data for the second site binders, giving valuable information for further optimization. 8
With full kinetic/affinity information for biomolecular interactions, we showed that SPR can be used to determine whether the analyte compound binds to one or more sites on the target. High-throughput experiments can be designed by using current SPR array platforms31,32 for compound/fragment library screening. Compared to common structural-based techniques (nuclear magnetic resonance, X-ray crystallography), SPR is very cost-effective in terms of protein consumption—only 15 µg of protein is required for achieving suitable immobilization levels and, depending on the protein properties, enabling the screening of hundreds of compounds in a single experiment. Considering there are many target proteins where the possibility of obtaining X-ray crystal structures is limited by protein availability or target accessibility (membrane proteins such as G-protein-coupled receptors33-35), SPR can be a powerful tool, providing information about compound binding and, with careful assay design, can help identify potential binding sites on targets and increase the likelihood of identifying compounds with unique properties.
The identification of kinase modulators with a dual-site mode of action, binding to both the ATP site and a second, allosteric site, creates an opportunity for novel kinase pharmacology. In oncology, two drug combinations of an ATP site inhibitor dosed with an allosteric site inhibitor have been shown to delay the emergence of drug-resistant mutations in Bcr-Abl 36 for the long-term treatment of chronic myeloid leukemia. Compounds that simultaneously bind to two distinct sites could provide a novel strategy for developing agents that delay the emergence of drug resistance. Compounds with dual-site modes of action have been identified here for ITK: Previously, the compound 4-FPP has been observed, by X-ray crystallography, to bind to both the ATP site and a C-terminal allosteric site in p38α MAP kinase. 37 The biosensor-based screening method reported here provides a systematic means to identify compounds that display similar dual-site or allosteric site modes of action in other members of the kinome.
Footnotes
Acknowledgements
We thank Marie Anderson, Alec Tucker, and Kimberly Skinner for their crystallization efforts and Iain Kilty for enzyme assay experiments. Thanks to Andrew Hopkins for comments on the manuscript.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The research was funded by Pfizer Global Research and Development. The authors received no financial support for the authorship and publication of this article.