
Abstract
Identifying chemical lead matter by high-throughput screening (HTS) has been a common practice in early stage drug discovery. Evolution of small-molecule library composition to include more drug-like molecules with desirable physical chemical properties combined with improving assay technologies has vastly enhanced the capability of HTS. However, HTS campaigns can still be plagued by false positives arising from nonspecific inhibitors. The generation of assay-ready plates has permitted an incremental advancement to the speed and efficiency of HTS but has the potential to enhance the occurrence of nonspecific inhibitors. A subtle change in the order of reagent addition to the assay-ready plates can greatly alleviate false-positive inhibition. Our case studies with six different kinase and protease targets reveal that this type of inhibition affects targets regardless of enzyme class and is unpredictable based on protein construct or inhibitor chemical scaffold. These case studies support a model where a diversity set of compounds should be tested first for hit rates as a function of order of addition, carrier protein, and relevant mechanistic studies prior to launch of the HTS campaign.
Introduction
High-throughput screening (HTS) is a commonly used practice in the early stages of drug discovery to identify novel lead matter that will become the basis of dedicated medicinal chemistry efforts. Advances in chemical informatics to efficiently estimate various physical and chemical properties have enabled research institutions to evolve their screening libraries to contain a higher proportion of drug-like molecules that obey the Lipinski rules. 1 Likewise, improvements in screening technologies such as increased precision and accuracy in small-volume dispensing have permitted rapid and robust ultra-high-throughput screening (uHTS) campaigns using low-volume and high-density microplates. Breakthroughs such as the advent of nanoliter acoustic dispensers have enabled the use of assay-ready plates (ARPs), wherein microplates are first seeded with nanoliter volumes of DMSO-solubilized test compounds to which assay reagents are subsequently added. In addition, ARPs provide higher flexibility to HTS labs in that assay plates can be generated in bulk in advance of several HTS campaigns. The ARPs can be stored frozen and used as needed for screening runs. Incorporation of ARP to our HTS workflow has dramatically improved screening time and assay statistics and has reduced reagent consumption and minimized cross-contamination of test compounds. However, we have found that our use of ARP can also result in high hit rates for several enzymatic screens. Further investigation found these higher hit rates to be associated with an increase in nonspecific inhibition.
The hit lists from HTS campaigns that are populated by nonspecific inhibitors (NSIs) often have uninterpretable structure–activity relationship (SAR) or, worse, SAR that can misdirect medicinal chemistry efforts. 2 Over the past decade, significant progress has been made, mostly by the academic research team of Shoichet, to clarify the mechanisms of nonspecific inhibition. Much of this work proved that promiscuous inhibitors physically interact with protein targets but do so by forming colloidal aggregates at micromolar compound concentration in an aqueous environment. 3 Upon binding to target proteins, these aggregates can partially denature or perturb the protein structure or conformation, thereby leading to nonspecific inhibition.4-6 Studies to identify these types of false-positive inhibitors during HTS triage have led to proposals such as the addition of detergent in an attempt to disrupt compound aggregate.7-9 The non-ionic detergent, Triton X-100, was proposed as the detergent of choice, but the concentration tolerated could vary with enzyme or receptor targets, different assay formats, and the chemical diversity of the library. 10 Another proposal to identify nonspecific inhibitors was to monitor the Hill slope because inhibition of this type is dependent on the critical aggregation concentration and is not expected to obey normal stoichiometric binding, thus leading to sharp concentration–response curves. 11 However, not all NSIs yield high Hill slopes, as shown in the case studies with β-lactamase; although the average Hill slope of compound aggregates was higher than that of true inhibitors, only 40% of aggregating compounds yielded Hill slopes greater than 1.5. 9 Some drug discovery researchers have suggested that nonspecific inhibitors could be easily removed by screening the compounds in the presence of an increased concentration of target protein.4,12 This approach, although useful, may be somewhat limited to assays that have a large dynamic range and are tolerant to an increase in enzyme concentration. In a related approach, inclusion of carrier proteins such as bovine serum albumin (BSA) or bovine gamma globulin (BGG) in biochemical assays serves a protective mechanism similar to increasing the enzyme concentration. However, BSA has discrete binding sites for anionic or hydrophobic molecules. 13 Thus, BSA may be less ideal for use in lead-finding HTS assays. In addition, detailed kinetic studies of inhibition mechanism and analytical approaches such as light scattering,14,15 surface plasmon resonance (SPR), 16 nuclear magnetic resonance (NMR), 17 and meniscus profile 18 have been applied post-HTS to help define which inhibitors appear to be nonspecific.
Although the molecular mechanism of nonspecific inhibition has been significantly clarified in recent years and both academic and drug discovery laboratories are well aware of the pervasiveness of the problem, there is no single satisfactory and prospective approach widely available to reduce the occurrence of nonspecific inhibition that frequently appears as false positives in HTS campaigns. The practice of experimentally identifying NSIs after the HTS run can be effective but often is inefficient because of the additional resources required (reagents, compounds, time). Thus, implementing preventative methods to avoid the activities of nonspecific inhibitors in the first place offers a better approach. Here we describe HTS campaigns against six different enzyme targets that yielded different susceptibilities toward nonspecific inhibition. The results from our studies concur with previous findings that promiscuous binding by compound aggregates (as detected by SPR) can emerge with one target class but not with another 16 and also provide several examples that extend the target-selective nature of nonspecific inhibition to different protein constructs of the same target. In addition, the susceptibility of each target to nonspecific inhibition was increased when using ARP, but subtle changes in the order of reagent addition to ARP greatly alleviated false-positive inhibition by NSIs. Our case studies also demonstrated that although carrier protein addition to assay buffer is an effective strategy to remove nonspecific inhibition when inclusion of detergents below the critical micelle concentration proved insufficient, the use of BGG instead of BSA offered a more inert system with respect to adventitious compound binding. Finally, these case studies exemplify the combination of some of these approaches with simple mechanistic characterization of the hits in HTS triage to increase confidence in the specificity of the compounds. Together, these findings document additional parameters of nonspecific inhibition in HTS campaigns that use modern methods of nanoliter– volume compound and reagent dispensation and exemplify the relative effectiveness of several practical strategies to minimize this nonspecific inhibition.
Materials and Methods
A summary of the assay buffer and detection technologies used for each target is found in Table 1 .
Assay Conditions for Each Enzyme
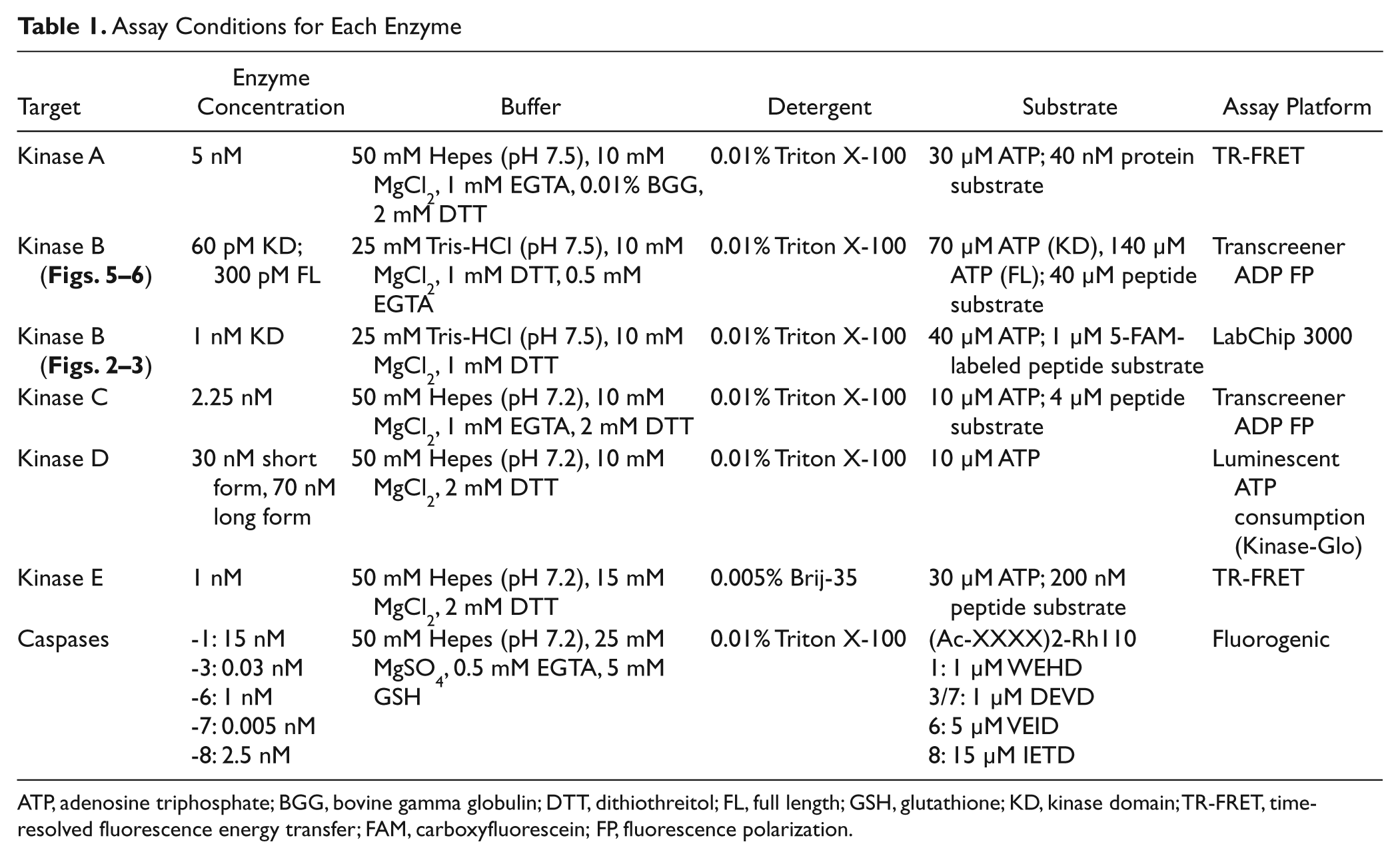
ATP, adenosine triphosphate; BGG, bovine gamma globulin; DTT, dithiothreitol; FL, full length; GSH, glutathione; KD, kinase domain; TR-FRET, time-resolved fluorescence energy transfer; FAM, carboxyfluorescein; FP, fluorescence polarization.
For typical HTS and IC50 triage, assays were performed in 1536-well MAKO plates (Aurora Biotechnologies, Carlsbad, CA) with 20-nL aliquots of compound stocks in 100% DMSO predispensed into plates using an Echo 550 acoustic dispenser (Labcyte, Sunnyvale, CA). The enzymatic reactions were initiated by addition of 2 µL of substrate or enzyme to 2 µL of substrate or enzyme preincubated with compound for 20 min at ambient temperature. BSA and BGG used in the assays were purchased from Sigma-Aldrich (St. Louis, MO) and were prepared as 10% (w/v) stock solutions while detergents were prepared v/v. The reagent dispense was conducted using BioRapTR noncontact dispensers (Beckman Coulter, Brea, CA). The reactions were quenched typically with an addition of 3 µL detection reagent. The plates were measured in a Wallac ViewLux (PerkinElmer, Waltham, MA) plate reader using a 400-nm dichroic mirror, a 340/50-nm excitation filter, and 618/8- and 671/8-nm emission filters for time-resolved fluorescence energy transfer (TR-FRET); an Alexa 594 dichroic mirror in combination with a 618/8 excitation filter with a polarizer and a 671/8 emission filter with parallel and perpendicular polarizers for Transcreener FP (Bellbrook Labs, Madison, WI) assay; or a clear emission filter for adenosine triphosphate (ATP) consumption (Kinase-Glo Promega, Madison, WI).
Non-ARP studies were typically performed in a 384-well format. Fluorogenic caspase assays shown in Figures 2 and 5 were carried out in 384-well black low-volume assay plates (PerkinElmer #6008260) in 12-µL reaction volumes containing enzyme, fluorogenic substrate, and inhibitor. All inhibitors were serially diluted in DMSO prior to dilution in assay buffer. Rhodamine-labeled bivalent tetrapeptide substrates were obtained from Anaspec (Fremont, CA). The plates were measured in an EnVision (PerkinElmer) instrument using excitation/emission wavelengths of 485/535 nm. Caspase assays shown in Figure 7 were similarly performed with the exception that compound was predelivered to 384-well plates in 100-nL volume in DMSO using the Echo 550 dispenser. Experiments for Kinase B were carried out in 384-well black assay plates (#MP101-1-PP; Matrical, Spokane, WA) in 20-µL reaction volumes containing enzyme, fluorogenic substrate, and inhibitor. The substrate and product were separated by capillary electrophoresis and detected using LabChip Caliper 3000 (Caliper Life Sciences, Hopkinton, MA). All inhibitors were serially diluted in DMSO prior to dilution in assay buffer and transfer to assay plate.
For all the concentration–response analyses, the data in each well were plotted as a function of inhibitor concentration, and the 50% inhibition (IC50) values were determined using a nonlinear least squares fit of the data to a four-parameter equation using Prism 5.0 software (GraphPad Software, San Diego, CA) or Screener 8.0 software (Genedata, Basel, Switzerland). Concentration–response curves for each inhibitor were normalized to zero and 100% activity based on controls with no enzyme or DMSO as sample, respectively. Each of the enzymes presented here shows little or no inhibition by DMSO even at the highest DMSO concentrations used in each experiment. In ARP enzyme-first conditions, the initial DMSO concentrations are 1% (v/v; typically 20 nL DMSO solvent in 2 µL).
Results
Effects of order of reagent addition on HTS hit rates
Because of the importance of detergents in eliminating promiscuous inhibition 11 by compound aggregation, it has been our standard practice to include detergent at concentrations approximately half of the reported critical micelle concentration (CMC) in our biochemical primary screen assays. In our recent HTS campaigns using ARP in low-volume 1536-well formats, however, we have encountered atypical high hit rates for test screens for a number of enzyme targets, which yielded low confirmation rates in either the same assay performed with intermediate compound dilution steps or with orthogonal assay technologies. Following further investigation, we found that for some assays, changing reagent order of addition to ARP from “enzyme first” to “substrate first” (without changing reagent volumes) can significantly lower the primary hit rate. The hit rate of a diversity set of 7000 compounds against Kinase A dramatically decreased in inhibition hit rate when the substrate solution was added to compounds in ARP prior to enzyme, despite inclusion of detergent (0.01% Triton X-100) and carrier protein (0.01% BGG) in the assay buffer for both methodologies. Two representative 1536-well ARPs subjected to each addition scheme illustrate the hit rate disparity ( Fig. 1A ). The order of addition was performed in parallel on ~2700 kinase-focused library compounds that contain known hinge binding motifs to compare the effect on the hit rate. The overall hit rate (defined as >50% inhibition at 5 µM compound concentration) for random HTS compounds decreased by 58% upon change in order of reagent addition to ARP, whereas only 9% of kinase-focused inhibitors were affected by the change of assay protocol. The relative loss of potency of these two compound classes with the change of reagent addition can be seen in Figure 1B . It is evident that “hits” with the greatest loss in their ability to inhibit Kinase A (>20% decrease in inhibition potency) largely represent compounds from the random HTS library, and the assumption that kinase-focused inhibitors will specifically bind to the ATP site of Kinase A suggests that the loss of potency of random HTS library compounds may be due to nonspecific inhibition. Our results from post-HTS mechanistic studies described in the next few sections support this hypothesis.
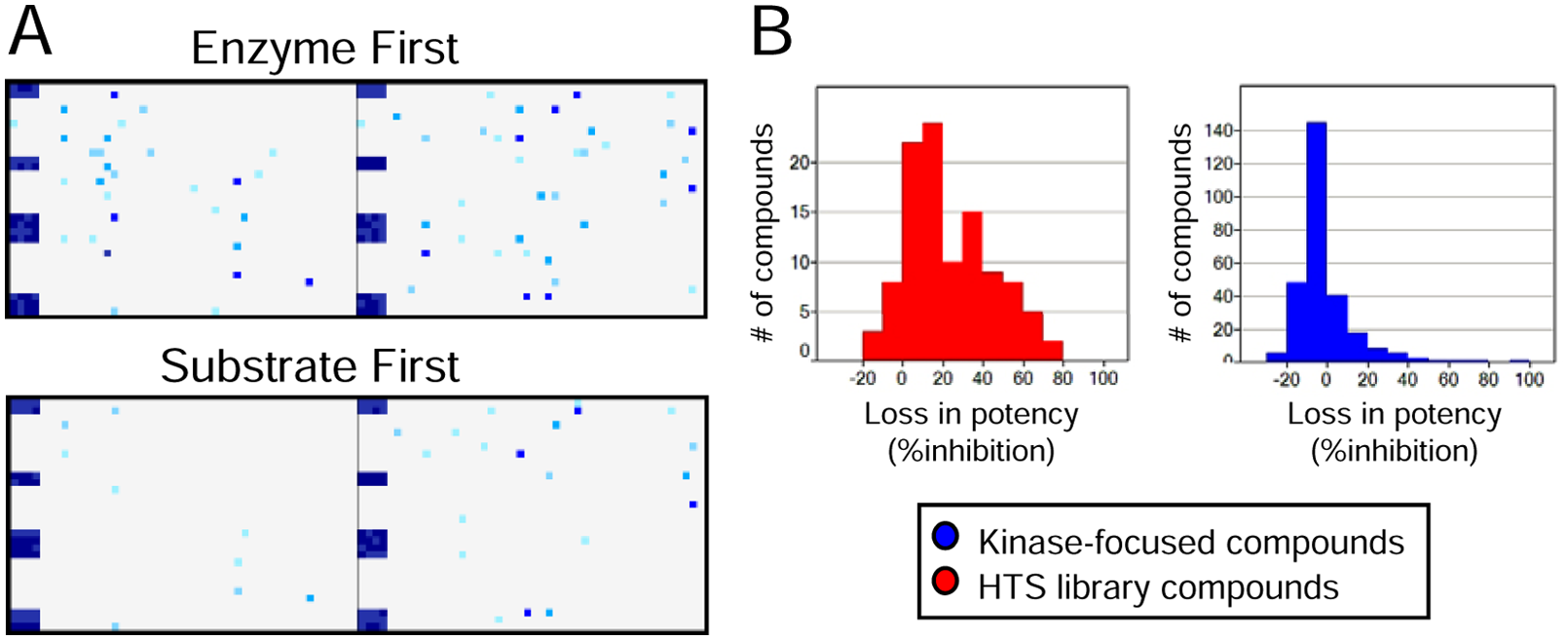
Effect of changing the order of reagent addition to assay-ready plates (ARPs) on hit rate of Kinase A. (
Concentration dependence of BGG for identifying and eliminating NSI
The effects of order of reagent addition on Kinase A in ARP suggested a significant population of NSIs and prompted us to investigate the effectiveness of carrier proteins to quench these persistent NSIs. The enzymatic activity of Kinase A is sensitive to the inclusion of high-concentration BGG (>0.01%), and we suspected that 0.01% BGG may not be sufficient to overcome nonspecific inhibition. To understand the effective concentration of the carrier protein in minimizing NSIs, we titrated BGG against a panel of nonspecific inhibitors identified from HTS against the cysteine protease caspase-6. Inclusion of BGG up to 0.3% had no effect on the activity of caspase-6, which exhibited a high degree of susceptibility to nonspecific inhibition. The 17 compounds from this panel were serially diluted in DMSO before being diluted in intermediate dilution plates with assay buffer containing 0%, 0.01%, 0.03%, 0.1%, or 0.3% BGG. Using a top compound concentration of 100 µM, the inhibitor potencies against caspase-6 were defined in a 384-well plate format. The inhibitory activity of 11 compounds was eliminated by the inclusion of 0.01% BGG (loss of inhibitory activity was determined as IC50 value greater than the highest compound concentration), whereas an additional 4 compounds were eliminated by 0.03% BGG, and 2 compounds lost inhibitory activity only at a BGG concentration of 0.1% (data not shown). The differential effect of BGG concentration on nonspecific inhibition highlights the variability of this type of inhibition and underscores a necessity of this target to incorporate a high enough concentration of carrier protein in an HTS campaign to effectively remove false inhibition. The concentration–response curves for one particular compound (compound 1) under each assay condition with caspase-6 illustrate this point ( Fig. 2A ). Although different methodologies were used for Kinase A and caspase-6, this result provided support to our hypothesis that 0.01% BGG may be insufficient for eliminating nonspecific inhibition for Kinase A. Conversely, a similar analysis was performed with the serine/threonine Kinase B and 20 different nonspecific HTS inhibitors tested with a maximal concentration of 30 µM. The assay was also performed in a 384-well format using intermediate compound dilution as described above. All compounds completely lost activity upon the inclusion of only 0.01% BGG, as illustrated in the concentration–response curve for compound 2 ( Fig. 2B ). Each NSI described here and throughout the article is defined based on at least one of the following criteria: promiscuous binding in SPR, aggregation in dynamic light scattering (DLS), loss of inhibition in high-concentration carrier protein, or Hill slope >1.5. None of the control compounds under any of these conditions was altered by the inclusion of BGG at any concentration tested (data not shown). Although further investigation is required to understand the disparity of BGG sensitivity for nonspecific inhibitors of caspase-6 compared with Kinase A or B, our results further emphasize the value of optimizing effective concentrations of carrier proteins, in addition to detergents, DMSO, or other additives, as well as order of reagent addition, prior to launch of an HTS campaign.
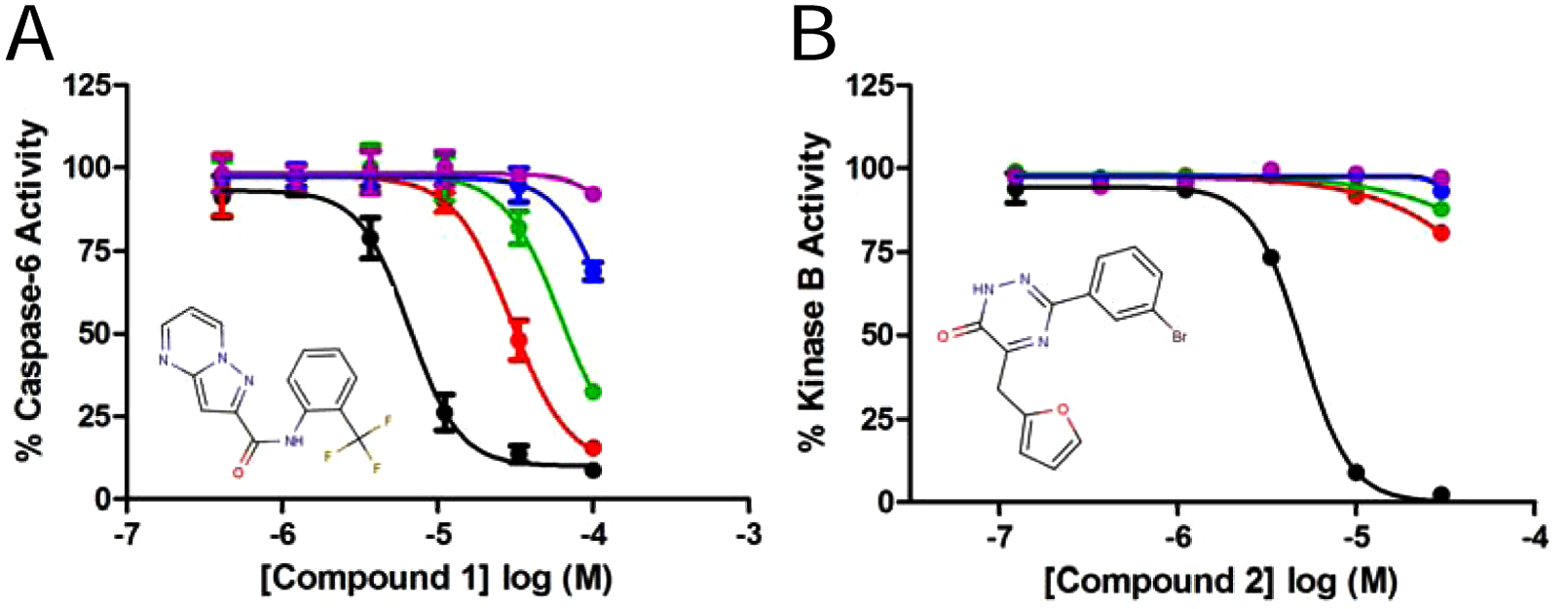
Concentration-dependent effect of bovine gamma globulin (BGG) in assay buffer on nonspecific inhibition of caspase-6 (
Specific inhibitor potency is decreased with inclusion of BSA in reaction buffer relative to BGG
For caspase-6 and potentially Kinase A, we demonstrated the benefit to include 0.1% BGG to effectively minimize nonspecific inhibition. Another commonly used carrier protein used for this purpose is BSA. However, as known specific small-molecule binding sites have been reported for BSA, the usage of this protein in micromolar concentration (~0.1%) may potentially yield false negatives by making them less available for interacting with the target protein. To indirectly assess the capacity of each carrier protein to bind and mask small-molecule inhibitors, we compared the potency of small panels of ATP site inhibitors against Kinase B with 0.1% BGG or 0.1% BSA present in assay buffer and using 384-well plates with intermediate compound dilution. ATP site inhibitors described here were determined as real inhibitors to their respective kinases by meeting at least one of the following criteria: active site binding observed by X-ray crystallography, potency shift under high ATP, or structural analog of known ATP site binder. Twenty-one of 30 ATP active-site inhibitors had a decrease in potency by more than twofold when BGG was replaced with BSA ( Fig. 3A ), presumably because of protein binding. 13 A similar analysis was performed for Kinase A except that the experimental design of this study was more challenging because of the lower tolerance of this enzyme to both BGG and BSA in reaction buffer. BGG could only be included in assay buffer up to 0.01%, whereas BSA was tolerated to only 0.05%. In this case, the potency of ATP site inhibitors was determined under these conditions with only 7 of 68 ATP active-site inhibitors decreasing in potency by more than twofold when 0.01% BGG is replaced with 0.05% BSA ( Fig. 3B ). Interestingly, the potency of two inhibitors increased by more than twofold when BGG was replaced with BSA. Thus, although BGG is the carrier protein of choice because of its relative inertness in binding to small molecules, the impact of BSA on the potency of true and specific inhibitors may vary depending on the chemical scaffolds of interest for a given target. For the simplicity of analyses, only BGG was used in mechanistic study for all our targets except in the case of Kinase A.
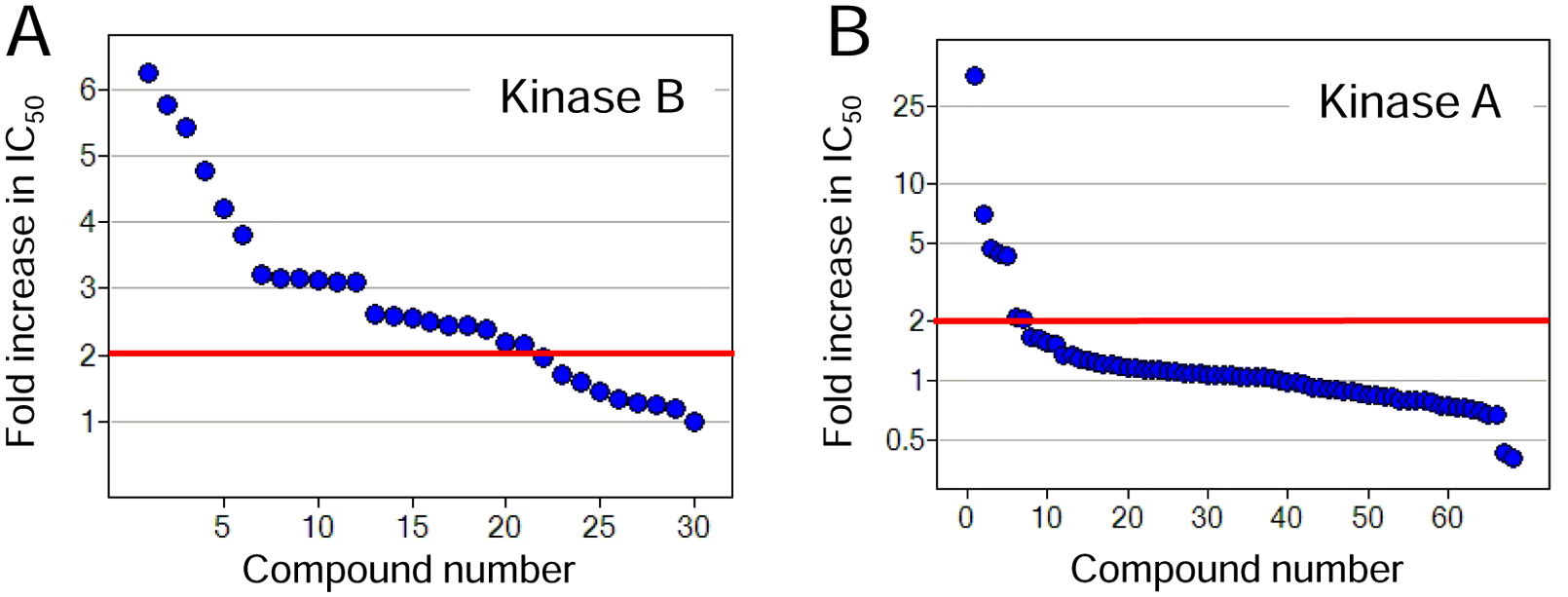
Effect of bovine gamma globulin (BGG) and bovine serum albumin (BSA) inclusion in assay buffer on inhibitor potency using a compound intermediate dilution protocol. (
Nonspecific inhibition is heterogeneous with respect to target
We tested the same diversity set of inhibitors described in Figure 1 for their activity against Kinase A with an increased amount of carrier, using 0.05% BSA in the assay buffer and substrate-first reagent order to ARP. For comparative purposes, the screening results of our diversity set for Kinase A when using 0.01% BGG with the enzyme-first protocol were correlated with the optimized protocol (0.05% BSA with substrate first; Fig. 4A ). When using this new protocol, the hit rate (>50% inhibition) for the HTS diversity set (red) decreased by 80% from 1.5% to 0.3%, whereas <10% of kinase-focused library hits (blue) were affected. These results illustrated the value of using a combination of strategies to minimize nonspecific inhibition and thereby effectively identify real hits in an HTS campaign.
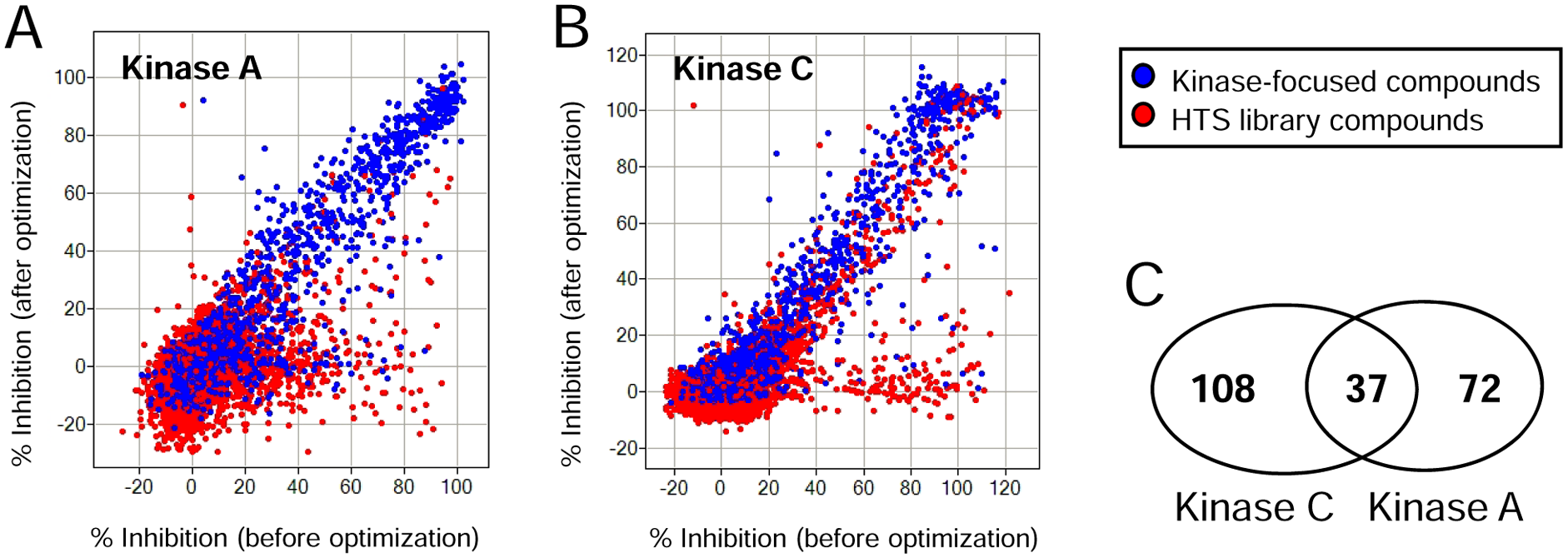
Hit rate comparison of a diversity set library when tested under original and optimized assay-ready plate (ARP) assay conditions. (
Taking into account order of reagent addition and inclusion of carrier protein as valuable methods to further reduce nonspecific inhibition in assays performed in ARP, we performed a similar analysis to compare NSI hit sets by screening this same diversity set of compounds at the same concentration against Kinase C. Both Kinases A and C are serine/threonine kinases, and both assays used similar HTS workflows (2 µL enzyme + 2 µL substrate in 1536-well plates). Like Kinase A, Kinase C also had a high hit rate (2.5%) for the validation screening set when carrier protein was absent from the reaction buffer with an enzyme-first protocol. Fortunately, the enzymatic activity of Kinase C is not sensitive to BGG. When 0.1% BGG was added in combination with the substrate-first protocol, the hit rate for Kinase C decreased by 60% from a hit rate of 2.5% to 1%, whereas there was only an approximately 10% decrease in the number of hits from the kinase-focused set ( Fig. 4B ). When the nonspecific inhibitors (defined as showing a loss in inhibition from >50% to within standard deviation of inactive compounds with the optimized protocol) were compared between Kinases A and C, only 37 of 217 total nonspecific inhibitors (17%) were in common between the two sets ( Fig. 4C ). This result provided further support to the conclusion that nonspecific inhibition by a given small molecule is highly target dependent, and it is not feasible to accurately predict the set of NSIs for one target based on the historical HTS data obtained with another target, even though the two targets belong to the same enzyme family and have similar HTS assay protocols (see Materials and Methods for details).
Nonspecific inhibition is enzyme isoform and protein construct dependent
The properties of an enzyme target that confer susceptibility to nonspecific inhibition have yet to be fully elucidated, yet it has been demonstrated that this effect is not limited to a particular enzyme class (e.g., Kinase A vs Kinase B vs caspase-6).2,4 Although high hit rates may be useful for diagnosing NSI, our collective HTS data have shown that hit rates can differ substantially even within the same enzyme class, in a manner that is not due to NSI. The challenge to predict enzyme susceptibility to nonspecific inhibition is illustrated by the diverse inhibitory profiles among caspase family members (-1, -3, -6, -7, and -8) in our first case study. Like all the members in the family, caspase-6 is a challenging target for drug discovery, partly owing to the presence of a reactive catalytic cysteine in the active site that is easily modified by electrophiles. Therefore, all compounds selected for additional studies against caspase-6 were chosen based on their lack of possible reactive moieties by employing stringent computational filters as well as demonstrating a lack of irreversible binding and sensitivity to various reducing agents (data not shown). A panel of 50 inhibitors that passed these requirements was tested in parallel for inhibition of caspase-6 as well as other family members caspase-1, -3, -7, and -8 using 384-well plates and intermediate compound dilution. There was a striking correlation between potency against caspase-6 and caspase-7 with near-absolute selectivity over the other highly homologous family members ( Fig. 5A ). As exemplified by the concentration–response curves for an example compound (compound 3) in Figure 5B , all enzymatic inhibition of caspase-6 and -7 by this set of compounds was eliminated by the addition of 0.1% BGG, suggesting that these compounds nonspecifically inhibit only two of these related enzymes. This variable sensitivity is apparently independent of enzyme concentration as well because caspase-3 was tested with 30 pM enzyme compared to 1 nM of caspase-6. These data again exemplify the target dependency of this type of inhibition and the value of verifying potential enzyme susceptibility prior to HTS initiation.
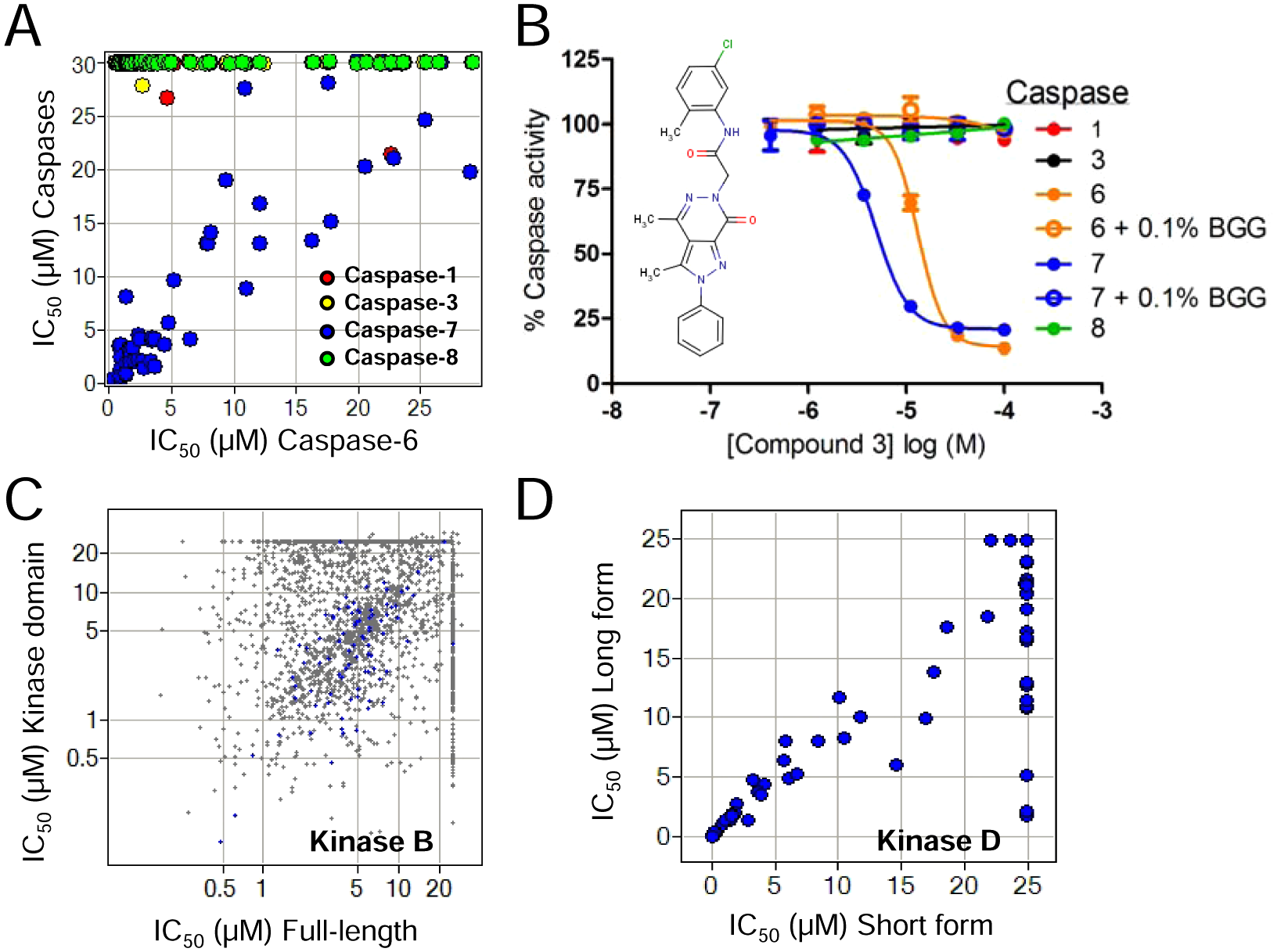
Nonspecific inhibition is heterogeneous with respect to protein class and protein construct. (
In addition to diverse profiles of nonspecific inhibition among members of the same family, we also have found protein construct–dependent NSI for a given enzyme target. Both full-length and the kinase domain protein constructs of Kinase B (a serine/threonine kinase) display similar specific activity and tool inhibitor pharmacology. When the two constructs were screened against a subset of library compounds in 1536-well plates using the same assay methodology in the absence of carrier protein and with the enzyme-first addition protocol to ARP, the overall hit rates were comparable, but only 54% of the total hits were common between the two ( Fig. 5C ). The compounds that were apparently unique inhibitors of each construct of this protein are unlikely kinase ATP site inhibitors as the active sites should be the same between the two. When the hits against full-length kinase were tested in the presence of 0.1% BGG, >95% of compounds (including those hits shared by kinase domain construct) completely lost their potency, suggesting that each construct of Kinase B was prone to additional different types of NSI ( Fig. 5C ). Nevertheless, 85 of the 88 compounds that retained inhibitory activity against Kinase B in the presence of BGG showed similar potencies between the two forms of enzyme in the absence of carrier protein. Thus, the authentic inhibitors behave largely as expected in the absence of carrier protein. Distinct protein constructs of different lengths were also generated for Kinase D, which is another serine/threonine kinase. In contrast to Kinase B, a longer 630–amino acid construct of Kinase D yielded a high hit rate with a significant number of inhibitors not active against a 354–amino acid kinase domain construct. The correlation of IC50s for each construct against a subset of these compounds in a 1536-well ARP format is shown in Figure 5D . When hits that were unique to the long enzyme were tested in the presence of 0.1% BGG, all inhibitory potency was lost, suggesting that the extra 276–amino acid extension on Kinase D may contribute to the increased nonspecific inhibition.
Noncompetitive mechanism of inhibition as a predictor of nonspecific inhibition for kinases
In addition to the usage of carrier protein and applying changes to the order of reagent addition, kinetic characterization of the mechanism of inhibition (MOI) can often provide evidence of nonspecific inhibition. 12 For example, when a kinase inhibitor exhibits apparent noncompetitive inhibition with respect to ATP, the inhibitory activity may often be due to either covalent or nonspecific inhibition, even though a true noncompetitive kinase inhibitor may be a desirable outcome. Two techniques are often employed to assess an apparent competitive mechanism of the compound hits in the HTS campaign: active-site ligand displacement and substrate titration assays.
In one case study targeting Kinase E, a screen was performed using 1536-well ARPs in the absence of carrier protein and with an enzyme-first protocol, which yielded a moderately high but not unreasonable 0.8% primary screen hit rate. The follow-up characterization using high and low concentrations of ATP surprisingly concluded that >80% of the hits were not ATP-competitive inhibitors (data not shown). For the entirety of Figure 6 , red circles are used to indicate those compounds whose MOI was defined as ATP-noncompetitive and blue circles as ATP-competitive inhibitors that yielded the expected shift between IC50 values with ATP in excess of its Km(app) relative to ATP at its Km(app). When compared in the presence and absence of 0.1% BGG, the majority of apparent ATP-noncompetitive inhibitors lost their inhibitory potency, whereas the reversible ATP-competitive inhibitors were not affected ( Fig. 6A ). The possibility of authentic noncompetitive inhibitors is of course a possibility. These data suggested that Kinase E was unusually sensitive to nonspecific inhibition in the primary screen assay and also that the true hit rate (excluding NSI) for this kinase was significantly lower than originally anticipated. Indeed, the primary screen hit rate with a modified protocol to include 0.1% BGG was less than 0.1% with >95% confirmed inhibitors demonstrating apparent ATP competitive inhibition. In another case, most apparent ATP-noncompetitive inhibitors of Kinase A—which did not show the expected IC50 shift upon an increase in ATP concentration—also lost all inhibitory potency when 0.01% BGG was replaced with a higher concentration of carrier protein (0.05% BSA; Fig. 6B ).
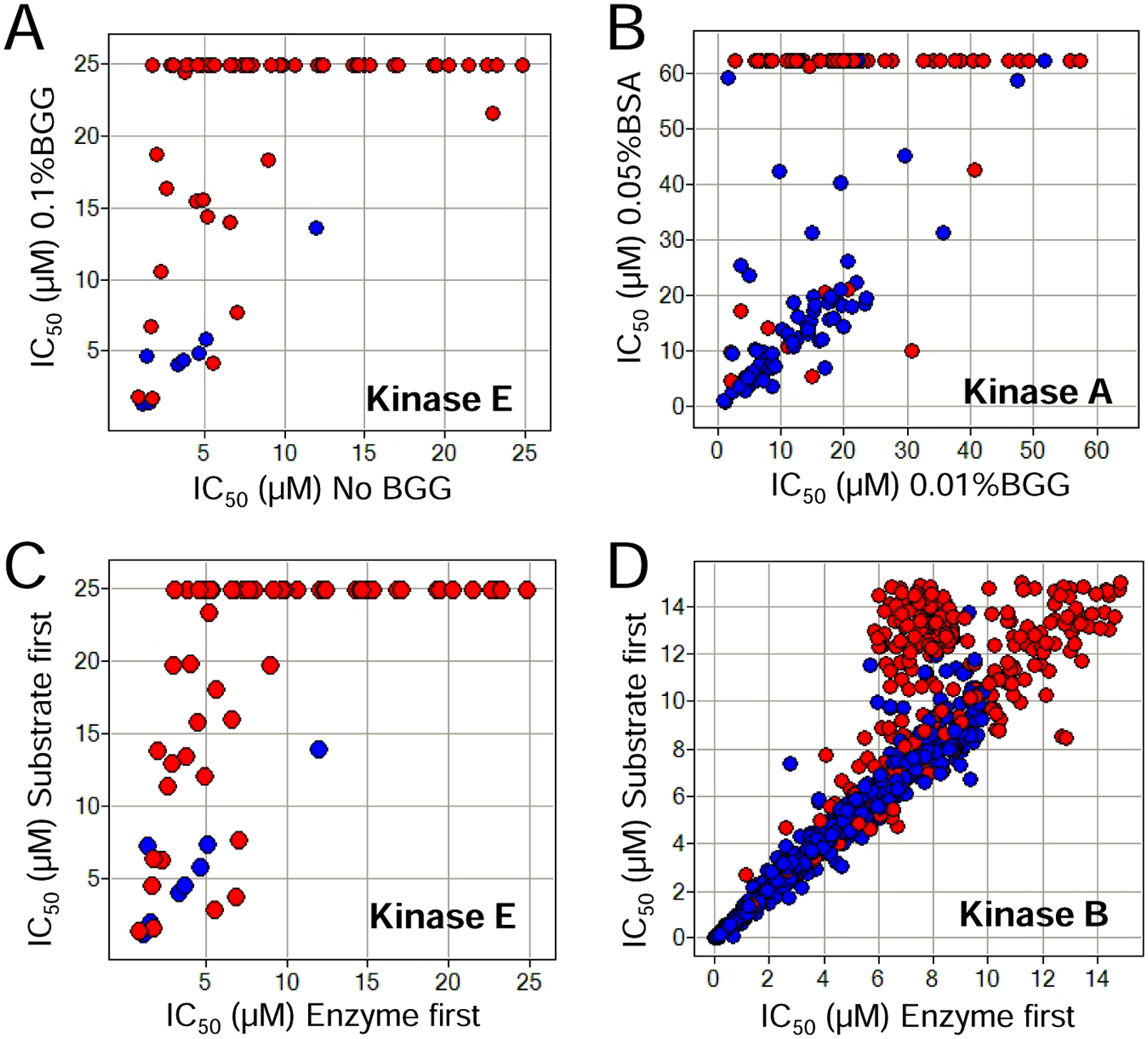
Nonspecific kinase inhibitors are frequently characterized mechanistically as noncompetitive with respect to adenosine triphosphate (ATP). (
Similar to the impact of carrier protein on NSI potency, the change of reagent order to ARP was also shown to preferentially decrease the potency of apparent ATP-noncompetitive inhibitors. Even in the absence of the carrier protein, by avoiding enzyme addition in the first step, 95 of 103 apparent ATP-noncompetitive inhibitors of Kinase E were shifted to weaker potency by more than twofold, whereas only 1 of 8 reversible ATP-competitive inhibitors yielded similar behavior ( Fig. 6C ). This result suggested an alternative option to minimize nonspecific inhibition if carrier protein is not well tolerated by the target enzyme or the assay detection. Similar results were observed with Kinase B ( Fig. 6D ), which was screened in the presence of 0.1% BGG. When IC50 values were compared between different orders of reagent addition, a significant number of ATP-noncompetitive inhibitors yielded weaker potency, whereas minimal impact was observed for the ATP-competitive inhibitors. Furthermore, those affected ATP-noncompetitive inhibitors also exhibited Hill slopes >1.5, consistent with nonspecific inhibition (data not shown). The ability of changing the reagent addition order to further eliminate potential nonspecific inhibitors was likely contributed by the preincubation of the compounds with the detergent and/or carrier protein in the absence of the target enzyme. These MOI correlation data are consistent with our hypothesis that the decrease in the hit rate shown in Figure 1 (by avoiding enzyme addition to ARP in the first step) is a result of reducing the number of nonspecific inhibitors (false positives) in the hit set without significantly reducing the identification of true inhibitors.
Reagent addition order to ARP alters NSI potency against caspase-6
To further elucidate the impact of reagent addition order on the potency of NSIs, we generated three copies of ARPs containing dose titrations of 13 caspase-6 HTS inhibitors verified to be NSIs based on the loss of potency in the presence of 0.1% BGG. Using BioRapTR liquid delivery instrumentation, caspase-6 enzymatic assays were performed in the absence of carrier protein by adding enzyme and substrate in the following schemes: (1) substrate added 20 min prior to enzyme, (2) enzyme added 20 min prior to substrate, and (3) simultaneous addition of enzyme and substrate. After 40 min of reaction progression, the apparent potency of each nonspecific inhibitor was measured, and the IC50s determined in schemes 1 and 2 were plotted relative to the IC50 determined with simultaneous addition of enzyme and substrate ( Fig. 7 ). Compound preincubation with caspase-6 resulted in an average 2.7-fold increase in potency relative to simultaneous enzyme/substrate addition. On average, the addition of substrate prior to enzyme had no effect on potency relative to the simultaneous addition scheme. These data support our assertion that for this target, enzyme preincubation with compound may contribute to an elevated number of NSIs in HTS campaigns.
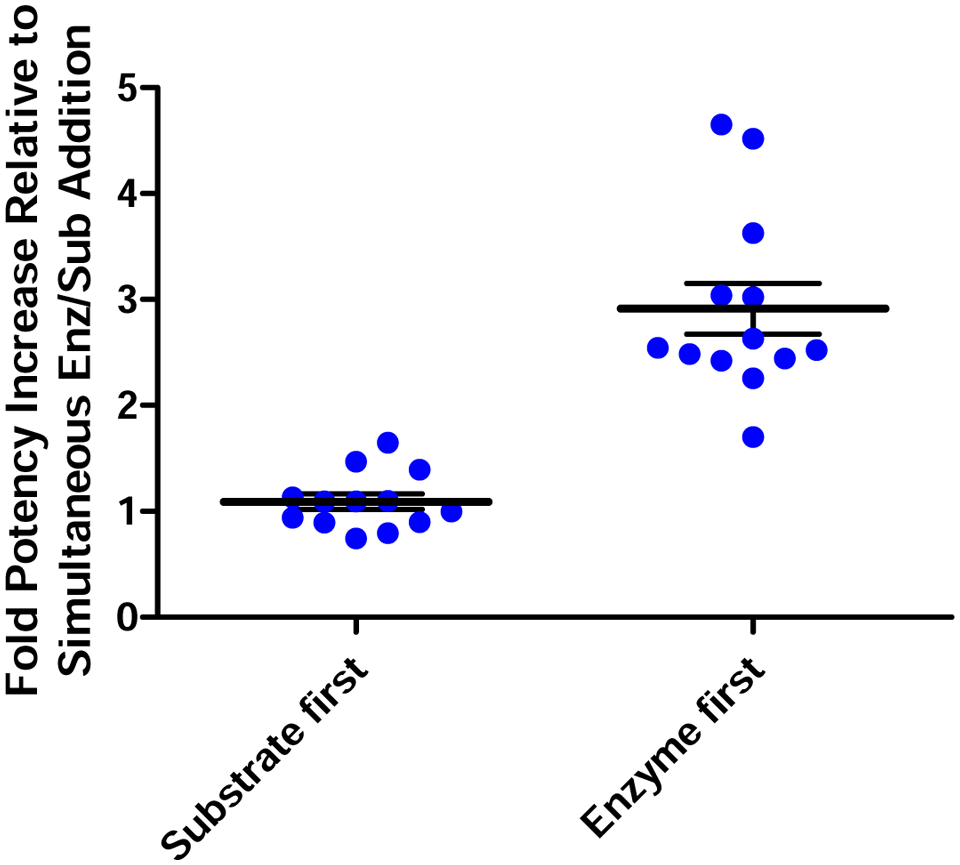
Effect of reagent addition ordering to assay-ready plates (ARPs) on nonspecific inhibitor potency against caspase-6. The potency of 13 nonspecific inhibitors is increased by 2.7-fold on average when enzyme-first protocols are used compared with simultaneous enzyme/substrate addition to ARP protocols. Nonspecific inhibitor potency is not affected when substrate-first ARP protocols are used.
Discussion
The continuous improvement in nanoliter dispensing precision and efficiency by either tip-based or contact-free acoustic dispense technologies has paved the way to generate assay-ready plates in routine HTS workflows. For a standard enzymatic assay, preincubation of compounds with enzyme has long been adopted to capture potential inhibitors with slow binding kinetics. With assay-ready plates containing compound in DMSO, a typical workflow is to add enzyme solution to resuspend compound prior to reaction initiation with substrate. In this paradigm, we have observed that adjustments to the order of reagent addition can have a profound effect on screening hit rate. With our ARP studies, we have observed the tendency of this enzyme-first workflow to yield higher hit rates than the reverse order of addition, and the additional hits were routinely characterized as nonspecific. Furthermore, the potency of nonspecific inhibitors was increased when applying enzyme-first protocols in lieu of substrate-first methods. This result is consistent with the commonly known time-dependent behavior of NSIs.4,19 In the referenced studies, activity was assessed after a 5-min incubation of enzyme with compound. Under circumstances of ARP use, the immediate dissolution of compound from DMSO into aqueous environment containing enzyme may not have permitted sufficient time for compound aggregates to be attenuated by detergent and/or carrier protein contained in assay buffer. Preliminary continuous kinetic data monitoring the initial rate of caspase-6 enzymatic activity in the presence of NSI support this hypothesis, in that inhibition of activity is evident even at the earliest measurable time points (data not shown). The 2.7-fold increase in potency after a 40-min reaction illustrated in Figure 7 indicates a degree of time-dependent onset of inhibition for these NSIs, but follow-up experimentation with assay systems capable of monitoring real-time fast kinetics would be required to truly dissect the temporal aspect of the initiation of nonspecific inhibition observed with the ARP.
In each of our case studies, the inclusion of detergent at a concentration half of the reported CMC was incapable of deterring all nonspecific inhibition, but the change of reagent addition order in ARP as well as the addition of carrier proteins to assay buffer proved instrumental in further minimizing their apparent potency. When BSA is used as a carrier protein, its known specific small-molecule binding sites may potentially complicate the interpretation of SAR by underestimating the potency of real inhibitors. Our indirect demonstration that BGG is less prone to interfere with real inhibitor activity relative to BSA has prompted us to favor use of this protein to minimize nonspecific inhibition when possible. More recently, other inert carrier molecules such as porcine collagen polypeptide fractions and casein-based blocking solutions have been commercialized for similar applications, and further study will be required to evaluate the advantages and limitations of these molecules in HTS. As illustrated in our case studies involving kinases, the combination of these preventative approaches with the mechanistic description of compounds functioning noncompetitively with ATP has also served as a valuable indicator of nonspecific inhibition during hit triage. The finding that the Kinase E hit set was populated so extensively with ATP-noncompetitive inhibitors further emphasizes the value of such mechanistic experimentation. The desire for highly selective kinase inhibitors as safe therapeutics continues to drive the search for unique noncompetitive binding modes that may offer greater selectivity. Directed attempts to identify real inhibitor binding outside of the active site will certainly be confounded by the presence of an overwhelming number of NSIs that appear to function in a similar mechanistic fashion.
Given the described challenges with identification of NSI in HTS campaigns, consideration of removing these compounds from the library is an understandable desire. However, unlike reactive molecules with known electrophilic, nucleophilic, or redox-prone functional groups that can be readily and discretely identified, a vast degree of heterogeneity for this type of nonspecific inhibition is both target and buffer dependent. From an HTS standpoint, the disparity we observe between nonspecific hit sets for Kinase A versus C clearly illustrates this point, which is consistent with the observation that promiscuous binding by compound aggregates can be target dependent. 16 Furthermore, this type of false-positive inhibition was not uniformly removed by a fixed concentration of BGG, suggesting that multiple forms of nonspecific inhibition may exist and may have different susceptibility to being quenched by the presence of BGG. Our collective studies also suggested that predicting the susceptibility to nonspecific inhibition of a given enzyme target is highly challenging. Comparing the members of the caspase family, we found that only caspase-7 shared a similar vulnerability to nonspecific inhibition as caspase-6. We cannot entirely rule out the possibility that other members of this cysteine protease family share a completely different set of NSIs because identical parallel screens were not performed on all enzymes. Furthermore, different constructs of the same protein can also play a significant role in the susceptibility to nonspecific inhibition. The kinase domain and full-length protein constructs of Kinase B were both susceptible to nonspecific inhibition, yet the majority within the NSI hit sets for each was distinct. In the case of Kinase D, an extra 276–amino acid extension appears to contribute to enhanced susceptibility to nonspecific inhibition. Our preliminary data for this enzyme have ruled out that protein folding and thermal stability of the different constructs is the predictor of NSI susceptibility (data not shown). Taken together, the data presented here highlight the importance of the order of reagent addition when using ARP and provide confirmation that successful hit triage requires a combination of strategies, including those outlined here as well as the proven techniques of monitoring inhibitor MOI and Hill slope. Given the indiscriminate nature of nonspecific inhibitors and the expanding use of nanoliter dispenser technologies across many aspects of drug discovery beyond HTS, further assessment of case studies across multiple target classes could be of value to the drug discovery industry.
Footnotes
Acknowledgements
We thank the entire baculovirus expression group and Protein Chemistry Department at Genentech, Inc. for cloning, expressing, and purifying many of the enzymes used herein, particularly Krista Bowman, Yvonne Franke, Mike Elliott, and Jiansheng Wu. We thank the Small Molecule Discovery Center at UCSF for kindly providing caspase-6. We also thank Rebecca Turincio, Steven Jones, and Richie Rodriguez for preparing ARP and running the uHTS screening system. We thank Anthony Giannetti for performing SPR and DLS experiments to confirm compound aggregation.