
Abstract
Epigenetic modification of DNA leads to changes in gene expression. DNA methyltransferases (DNMTs) comprise a family of nuclear enzymes that catalyze the methylation of CpG dinucleotides, resulting in an epigenetic methylome distinguished between normal cells and those in disease states such as cancer. Disrupting gene expression patterns through promoter methylation has been implicated in many malignancies and supports DNMTs as attractive therapeutic targets. This review focuses on the rationale of targeting DNMTs in cancer, the historical approach to DNMT inhibition, and current marketed hypomethylating therapeutics azacytidine and decitabine. In addition, we address novel DNMT inhibitory agents emerging in development, including CP-4200 and SGI-110, analogs of azacytidine and decitabine, respectively; the oligonucleotides MG98 and miR29a; and a number of reversible inhibitors, some of which appear to be selective against particular DNMT isoforms. Finally, we discuss future opportunities and challenges for next-generation therapeutics.
Introduction
For nearly 60 years, it has been recognized that cellular inheritance is controlled by DNA replication during cell division. DNA mutations and gene rearrangements were later discovered to be the driving force behind the evolution of unique organisms. Despite having identical DNA sequences, cells within an organism differentiate into a wide variety of tissues and cell types. The explanation for this phenomenon was built on a proposal that DNA is organized with a complex of proteins into nucleosomes: DNA wrapped 1.67 turns around a histone octamer comprising two molecules each of the common histones H2A, H2B, H3, and H4.1–4 The discovery that reversible molecular modifications of histones and DNA control gene expression introduced a novel paradigm known as epigenetics.
Recent studies indicate that epigenetic silencing has similar effects as DNA mutations in tumorigenesis. 5 In contrast to DNA mutations, epigenetic silencing is a reversible stepwise process, with various levels of control. Transient histone modifications (phospho, methyl, acetyl, ubiquityl, and sumoyl) and more stable DNA methylation lead to the compaction of nucleosomes and formation of transcriptionally inaccessible heterochromatin ( Fig. 1A ), which needs to be faithfully replicated during cell division to maintain repressive marks ( Fig. 1B ). Reactivation of genes is also stepwise, resulting in the formation of readily accessible euchromatin. During epigenetic remodeling, many components are required to modify or bind DNA and histones. These concepts have been reviewed extensively.6–14 This review focuses on the role of human DNA methyltransferases (DNMTs) in cancer as well as the therapeutic potential and challenge of inhibiting DNMTs.
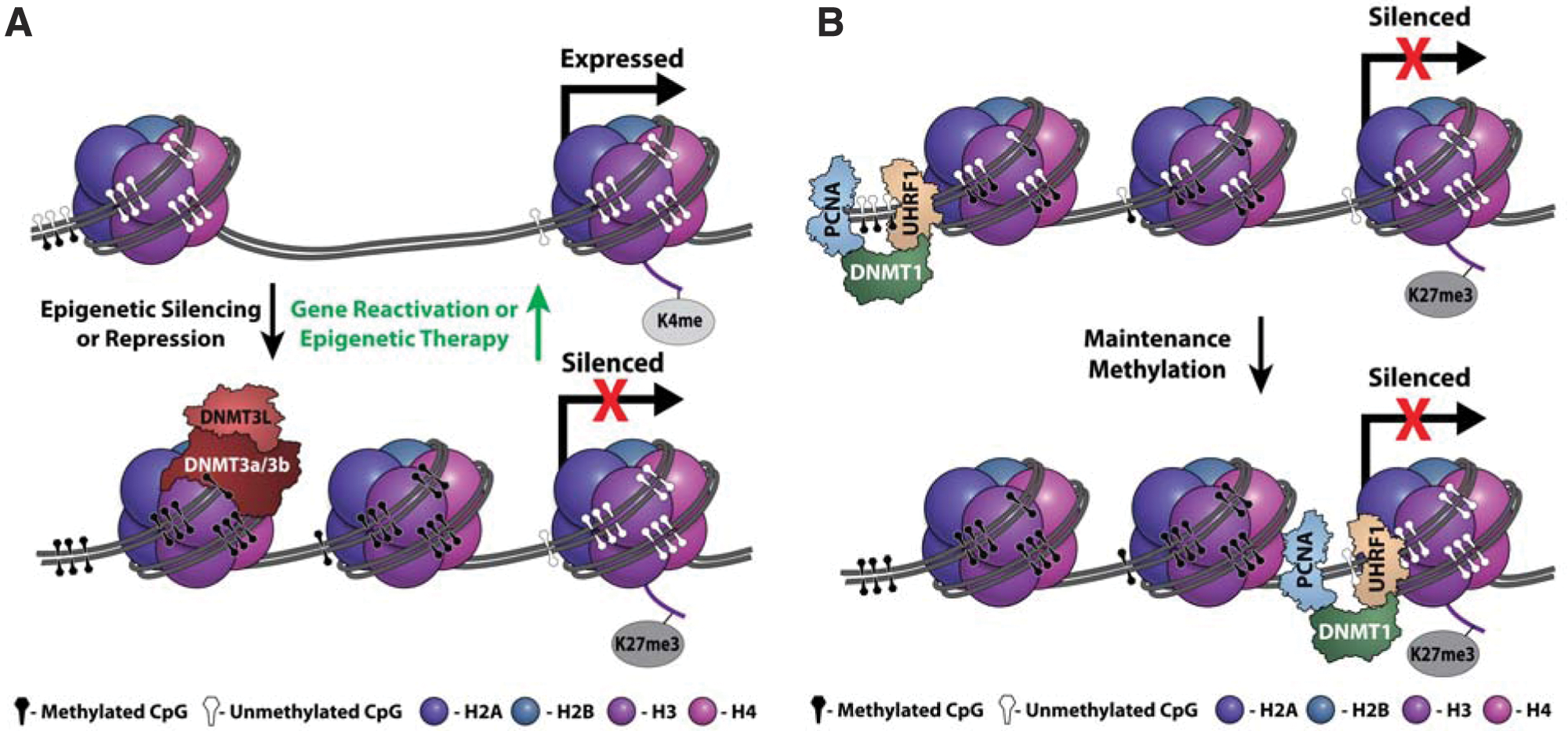
(
DNA methyltransferases
DNA methyltransferases transfer a methyl group from S-adenosyl-methionine (SAM) to the C5 position of the pyrimidine ring of cytosine residues in genomic CpG dinucleotides. CpG dinucleotides are preferentially located in specific regions of the genome, including clusters in gene promoters called CpG islands. 15 CpG methylation is crucial for silencing retrotransposon element sequences (i.e., LINE-1 or Alu repeats), X chromosome inactivation, imprinting, regulating gene expression, and cellular transformation.14–16
There are five DNMT isoforms—DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L—but only DNMT1, 3a, and 3b methylate DNA. DNMT1 is a maintenance methyltransferase, copying methylation marks after DNA replication. This is evident by a preference for hemimethylated DNA as its substrate17–19 and association with two proteins that assist in methylation maintenance during replication: proliferating cell nuclear antigen (PCNA) and ubiquitin-like plant homeodomain and RING finger domain-containing protein 1 (UHRF-1).20,21 It has been shown that the PCNA binding domain of DNMT1 enhances but is not required for DNA methylation maintenance. 22 In contrast, UHRF-1 is required to maintain DNA methylation by recruiting DNMT1 to hemimethylated DNA near replication forks and damage sites. 21 DNMT1 and UHRF-1 deficiency in mice are embryonically lethal23,24 and result in global loss of DNA methylation in embryonic stem cells, embryos, and mouse embryonic fibroblast (MEF) cells.25–27 Complete knockout of DNMT1 is catastrophic for the HCT-116 human colon cancer cell line.28,29
Despite sequence similarity to the DNMT family, DNMT2 does not methylate DNA but instead methylates a specific cytosine in tRNAAsp and has been alternatively named tRNA aspartic methyltransferase 1 (TRDMT1).30,31 DNMT2 null mice are viable and fertile, 30 and it remains unclear whether DNMT2 plays a role in human disease. 31
DNMT3a and DNMT3b are de novo methyltransferases, initiating DNA methylation marks on unmethylated DNA, and are critical for development and cellular differentiation.32–34 DNMT3a-deficient mice display organ defects and lethality within weeks after birth with only minor DNA methylation changes. 32 DNMT3b-deficient mice are embryonic lethal, suggesting a more significant role of DNMT3b in early development compared to DNMT3a. 32 DNMT3b-deficient mice exhibit drastic defects in minor satellite repeat methylation but only minor changes at other common methylation sites. 32 Mice deficient for both DNMT3a and DNMT3b are embryonic lethal, and DNMT3a–/– DNMT3b–/– mouse embryonic fibroblasts display a dramatic loss of de novo methylation activity and changes to global methylation despite the presence of DNMT1.29,32 It has also been shown that DNMT3a/3b can cooperate with DNMT1 for maintenance methylation.18,35–37
Further differences between DNMT3a and 3b functions are evident in conditional germ cell knockout models. DNMT3a and DNMT3b conditional knockout mice are viable, but only offspring from DNMT3a conditional knockout mice die in utero and lack methylation-dependent imprinting, the silencing of maternal or paternal alleles during gametogenesis. 38 Thus, although DNMT3a and DNMT3b have overlapping de novo methylation functions, it is clear that DNMT3a is critical for imprinting and germ cells, whereas DNMT3b is more essential for early somatic development.
The remaining DNMT isoform, DNMT3L, shows high homology to DNMT3a and DNMT3b but is catalytically inactive. DNMT3L knockout mice are viable, but both sexes are sterile, and female DNMT3L knockout mice do not produce viable offspring when crossed with wild-type mice. 39 Oocytes from DNMT3L-deficient female mice and their heterozygous progeny lack maternal methylation imprints, whereas male germ cells are deficient in paternal methylation imprints.39,40 Because DNMT3L is catalytically inactive and the other DNMT3 family members are de novo methyltransferases, genetic models were evaluated and revealed a cooperative role of DNM3L and DNMT3a/3b for methylation imprinting. 41 Later studies confirmed that DNMT3L binds to and enhances DNMT3b and DNMT3a enzymatic activity.42,43
DNMT mutations
Mutations in all three catalytically active human DNMT isoforms have been described. Recently, DNMT1 mutations were identified and linked to a form of hereditary sensory and autonomic neuropathy type 1 (HSAN1) with dementia and hearing loss. 44 The heterozygous mutations occur in the DNMT1 nuclear targeting region, which is known to be critical for DNMT1 activity. 45 Despite the reduction of activity and measurable changes in global methylation, HSAN1 patients with DNMT1 mutations do not show an increased propensity for cancer, countering mouse model evidence that will be discussed later in the review.
DNMT3b mutations have been well characterized as the cause of immunodeficiency, centromere instability, facial anomalies (ICF) syndrome. 46 ICF patients have compound, biallelic mutations that reduce DNMT3b methyltransferase activity. Due to the rare occurrence of ICF syndrome (fewer than 50 patients reported worldwide since 1970) and the observed residual methyltransferase activity in the mutated forms of DNMT3b, it is believed that humans with complete functional knockout of DNMT3b may die at birth, similar to the phenotype observed in knockout mice. 47 On a cellular level, ICF DNMT3b mutations result in satellite hypomethylation and juxtacentromeric rearrangements of chromosomes 1, 9, and 16. There has been no observed increased risk of cancer with ICF patients, most likely due to a shortened life span because of immunodeficiency, but also possibly due to an upregulation of DNA damage response elements that prevents more rampant genomic instability. 48 The distinction could be important since the latter scenario would indicate that therapies targeting DNMT3b may enhance radiation-induced growth inhibition.
Finally, heterozygous DNMT3a mutations have been reported in a subset of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) patients and correlate with poor prognosis compared to patients lacking DNMT3a mutations.49,50 AML patients with DNMT3a mutations have residual wild-type expression, do not display global methylation changes, and do not appear to have increased genomic instability based on a normal karyotype and lack of additional mutations, making the oncogenic mechanism unclear. Although more work needs to be done to explore the function of DNMT3a in leukemia, a recent study confirmed the presence of DNMT3a mutations in subtypes of AML patients and found methylation and gene expression changes in the HOXB locus, which has been implicated in oncogenesis.51–53 Thus, of the known DNMT mutations in humans, only DNMT3a mutations have been implicated in an increased risk for cancer, whereas targeting DNMT1 and DNMT3b for oncological therapy may have less inherent risk of tumorigenesis.
DNA methyltransferases and cancer
DNA methylation profiles in cancer cells are unique from normal, differentiated cells.54,55 In cancer cells, widespread hypomethylation occurs throughout the genome, and enriched hypermethylation is focused in CpG islands.14,54,55 CpG islands are located in the promoters of tumor suppressor genes, including some of the most commonly mutated genes in cancer such as retinoblastoma (Rb), von Hippel-Lindau (VHL), cyclin-dependent kinase inhibitor 2A (CDKN2A, p16), adenomatous polyposis coli (APC), and others listed in Table 1 .
Tumor Suppressor Genes Silenced by Hypermethylation in Various Cancers and Their Roles in Transformation
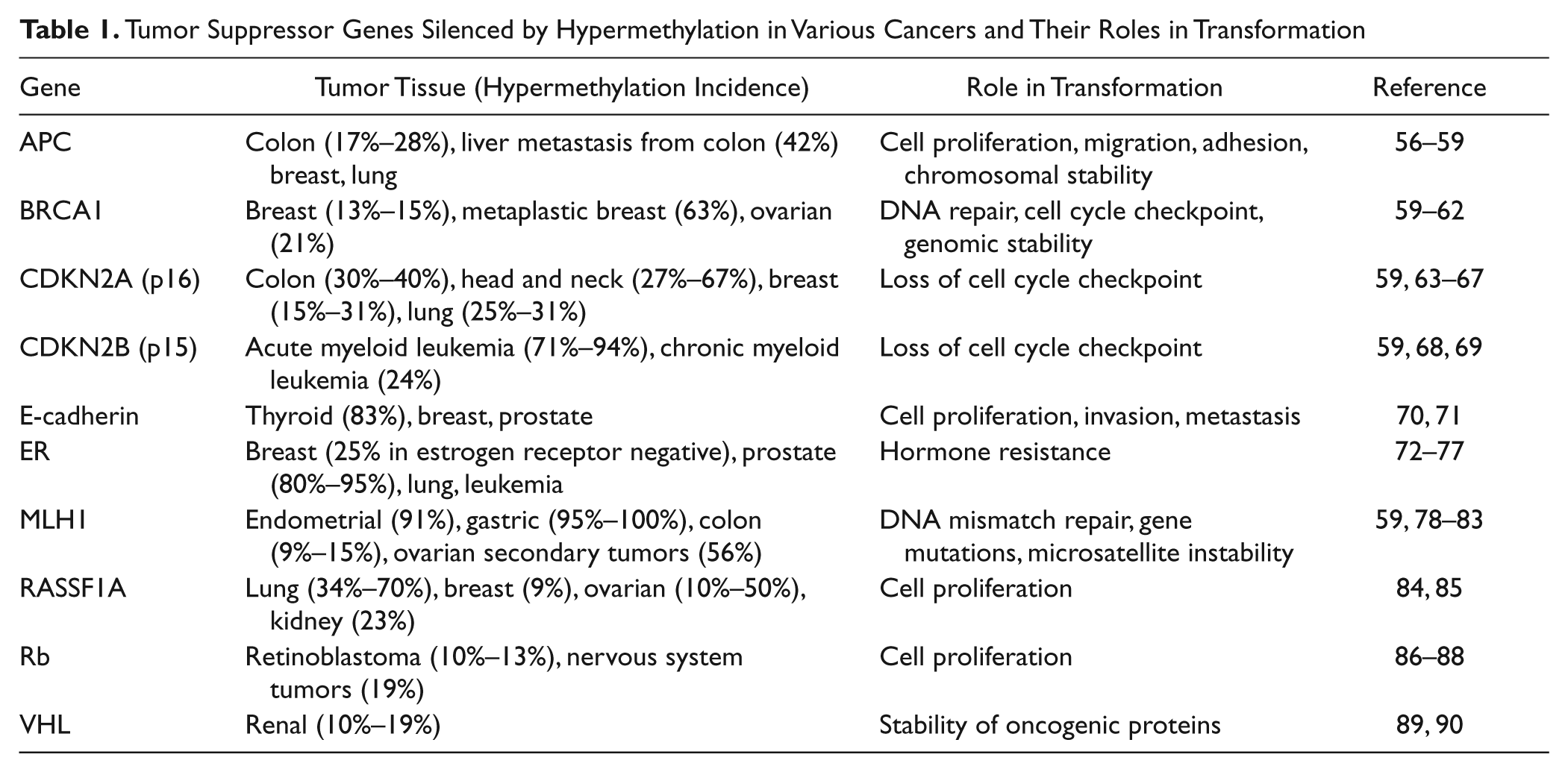
Silencing of tumor suppressor genes through DNA methylation has been proposed as a “second hit” for cellular transformation, equivalent to mutations or translocations. 5 Related to this, methylation-induced silencing occurs in genes relevant to a particular cancer progression: estrogen receptor (ER) in breast cancer, breast cancer type 1 susceptibility protein (BRCA1) in breast and ovarian cancer, mutL homolog 1 (MLH1) in colon cancer, and so on. 91
Studies in mice with APC deficiency have revealed a role for DNMTs in the formation of colon tumors. APCmin mice develop intestinal neoplasias, but APCmin/DNMT1+/– mice exhibit reduced frequency of intestinal neoplasia. 92 APCmin/DNMT3b–/– mice also develop fewer intestinal neoplasms but develop the same amount of early stage adenomas as APCmin mice, implicating DNMT3b for later stage tumor growth. 93 Overexpression of DNMT3b, but not DNMT3a, in APCmin mice promoted tumorigenesis and increased methylation of the tumor suppressor genes SFRP2, SFRP4, and SFRP5 but did not alter methylation states for APC, MLH1, or p16, suggesting that gene-specific methylation occurs during tumorigenesis. 94
Mounting evidence suggests that microRNAs (miRNA), small noncoding RNAs that alter gene expression, can be regulated by DNA methylation and can indirectly regulate cancer-relevant gene expression. 95 One particular miRNA, mir-124a, is hypermethylated in acute lymphoblastic leukemia (ALL) patients and reexpressed following treatment with a hypomethylating agent. 96 In addition, reexpression of mir-124a correlated with silencing of its target CDK6, a protein involved in the Rb pathway. 97 Thus, methylation changes may result in a cascade of downstream effectors that may alter cancer progression.
DNMT1 and DNMT3b are overexpressed and amplified in numerous cancers. 98 Initial studies with DNMT1 knockout in HCT-116 cells revealed a cooperative role for DNMT3b because only DNMT1/DNMT3b double knockouts displayed altered methylation levels and slowed proliferation. 35 Tumor xenograft growth is also reduced in the double knockout cells when compared to parental cells. 99 It was later discovered that the original DNMT1 knockout was a hypomorph and that complete DNMT1 knockout is catastrophic for the HCT-116 cancer cell line.28,29 Interestingly, DNMT1-deficient mouse embryonic fibroblasts require multiple cell divisions for apoptosis to occur, suggesting that cancer cells may be more reliant on DNMT1 activity than normal cells for survival. 26
Alternative splice variants of DNMT3b also are upregulated in cancers and alter DNA methylation patterns despite the fact that some are catalytically inactive.100–103 It is unclear how these truncated DNMT3b isoforms affect normal DNMT3b function and contribute to oncogenesis. Perhaps overexpression of catalytically inactive DNMT3b plays a role in the global hypomethylation phenotype observed in cancer cells. 14
A concern of inhibiting DNMTs is on-target toxicity and oncogenic events that may be the result of retrotransposon reactivation, genomic instability, or spontaneous immortalization.104–106 These and other studies have indicated that low levels of DNMT1 lead to hypomethylation, even lower levels induce genomic instability, 107 and complete knockout is lethal. 108 Although these observations are alarming, studies with MDS patients treated with nucleoside analog hypomethylating agents have not revealed an increase in chromosomal instability above normal levels, 109 and normal cells with lower proliferation capacity are less sensitive to hypomethylating agents.110,111 In addition, the potential oncogenic effects of hypomethylation may actually sensitize cancer cells to apoptosis through genomic instability or enhance immune-targeted cell death through expression of tumor antigens. 112
The following sections summarize the current DNMT targeting agents that have been tested preclinically and those that are approved commercially. Although two DNMT inhibitors are currently approved to treat MDS, there are potential advantages for creating new generations of inhibitors to improve drug properties, eliminate the need for DNA incorporation, and target solid tumors. The key will be to find the appropriate therapeutic window and identify combinations that may limit toxicity and avoid oncogenesis.
Past and Current Approaches to DNMT Inhibition
Nucleoside analogs
Historically, the first compounds that demonstrated DNA hypomethylation activity in cells were cytidine analogs: 5
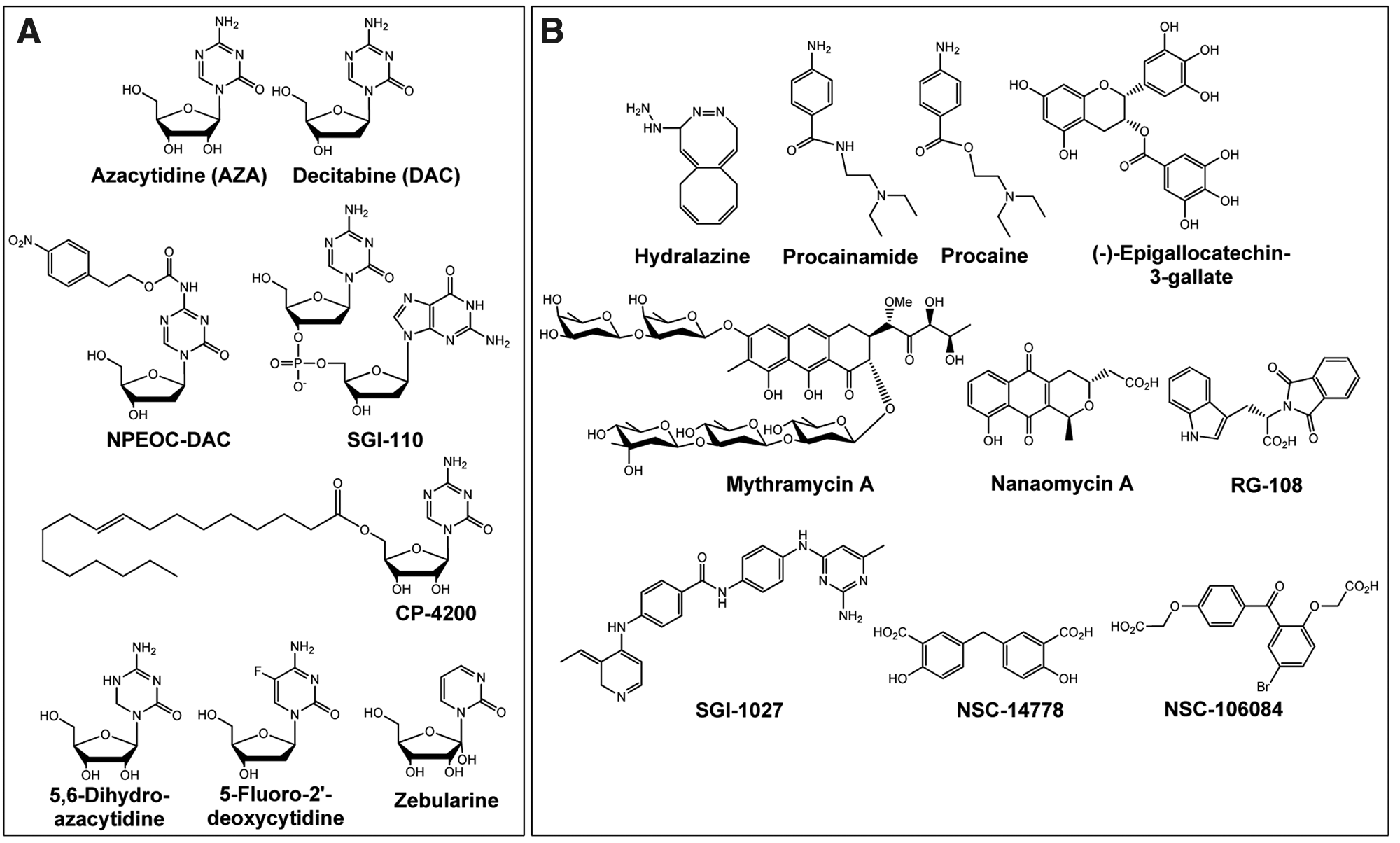
Structures of compounds with reported DNA methyltransferase (DNMT) inhibitory activity. (
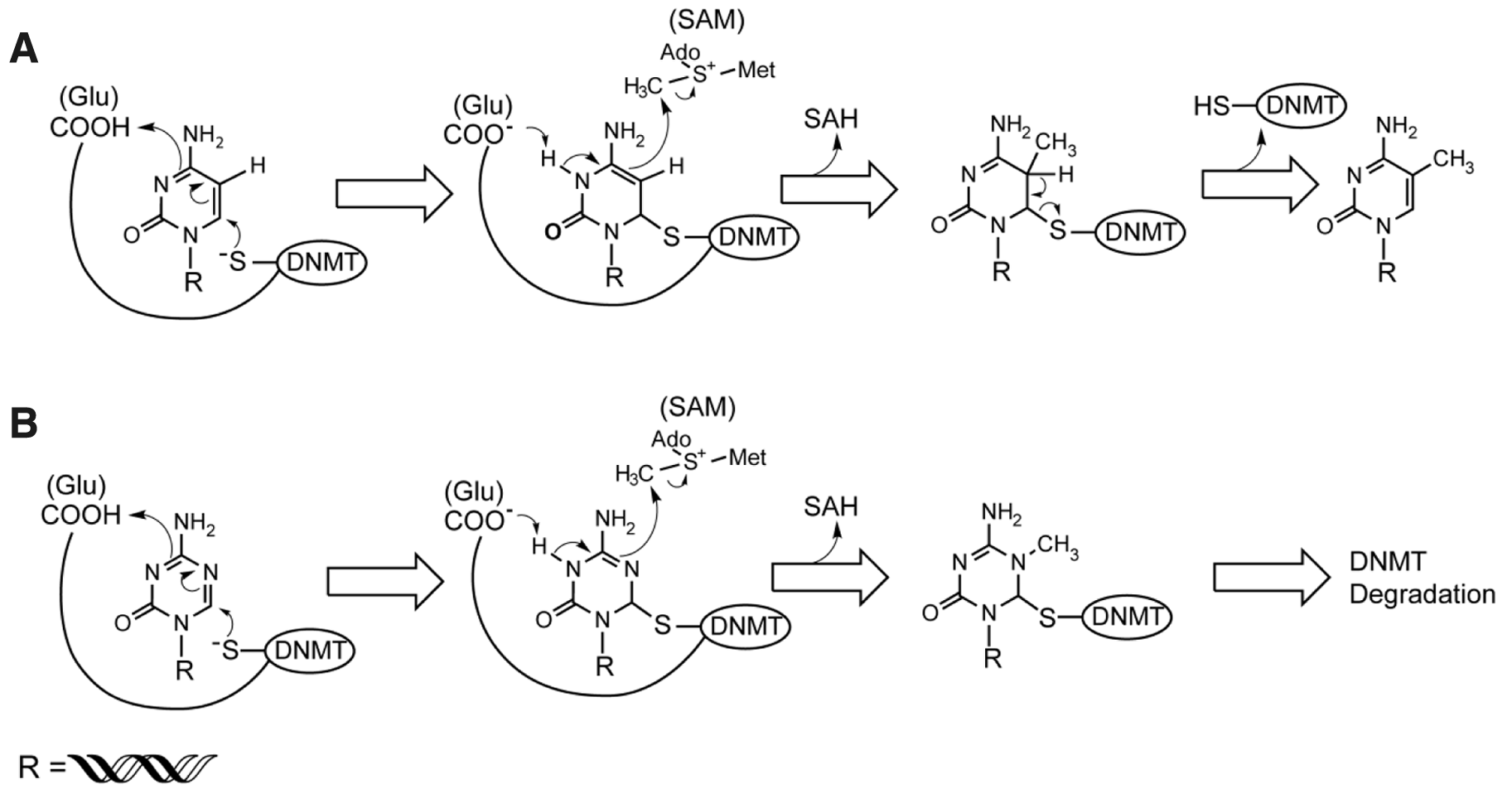
(
The potent DNA hypomethylation activities of DAC and AZA disappear at higher concentrations (~3–10 µM).116,117 The U-shaped dose–response curve for hypomethylation reflects cytotoxicity that is unrelated to DNA methylation, resulting in diminished DNA incorporation of DAC and AZA. 116 Most of the early clinical trials of AZA and DAC used dosages that were based on maximum tolerated doses that may not have effectively decreased DNA methylation.119–121 The early clinical trials were designed to test the potential cytotoxic effect of the inhibitors rather than whether DNMT inhibition would be an effective cancer therapeutic modality.
In the past decade, clinical trials in MDS and AML have employed low doses of AZA and DAC along with treatment schedules that allow for efficient DNA incorporation with minimal toxicity and have shown clear clinical benefits. AZA was approved by the U.S. Food and Drug Administration (FDA) in 2004 for the treatment of MDS, based on a primary phase III trial where patients treated with AZA showed an improvement in objective response rates and a reduction in transfusion requirements relative to best supportive care. In the primary AZA registration study, the response rate in the AZA group was 16% (6% complete response and 10% partial response), and there were no responses in the best supportive care arm.122–124 A subsequent European phase III trial showed significant improvement in median overall survival: 24.5 months with AZA versus 15.0 months with conventional care regimens. 125 AZA has been granted marketing authorization by the European Commission for patients with high-risk MDS and AML.
DAC was also approved for MDS treatment in 2006, based on an objective response rate and transfusion independence in a randomized phase III study where DAC was compared to best supportive care. In the DAC registration study, patients in the DAC treatment arm showed an objective response rate of 16% (15% complete response rate and 1% partial response). 126 Using preclinical findings that showed optimal DNA hypomethylation with low-dose prolonged exposure to DAC, 127 subsequent clinical trials showed significantly improved results, including 39% complete responses, 128 and an overall improvement rate of 51%. 129 Similar results were observed in a recent phase II study of DAC in older patients with AML, including a 27% complete response rate. 130 The clinical benefits of DAC appear to be mechanism specific, as clinical response in MDS has been shown to correlate strongly with DNA hypomethylation. 131
On the basis of success of AZA and DAC in hematological disease, clinicians are now using the low-dose, prolonged exposure treatments pioneered in MDS clinical trials in a solid tumor setting. A recent phase I study using DAC in combination with carboplatin in platinum-resistant ovarian cancer showed promising results, including complete responses and stable disease as well as DNA hypomethylation in peripheral blood mononuclear cells. 132 A phase II combination study of AZA and the histone deacetylase inhibitor, Entinostat, in advanced non–small cell lung cancer is also under way, with early results indicating that 30% of the first 28 patients on the trial are responding to treatment. 133
Despite the advantages of AZA and DAC, significant challenges stem from the instability of both compounds in aqueous solutions as well as in vivo deamination by cytidine deaminase. Efforts to circumvent problems of metabolic instability have yielded two different analogs of DAC: NPEOC
In addition to improving the stability of the azanucleosides, efforts are under way to improve cellular delivery. CP-4200 is an elaidic acid ester analog of AZA designed to render the drug less dependent on conventional nucleoside transport systems and has shown superior efficacy to AZA in an orthotopic ALL mouse tumor model. 138
Other nucleoside analogs
In addition to 5-Aza-C and 5-Aza-CdR, other cytidine analogs have been shown to inhibit DNMT activity once incorporated into DNA. Among these is 5,6-dihydroazacytidine (5,6-dihydro-AZA), which has demonstrated DNA hypomethylation activity in cells, albeit with 10-fold less potency than AZA. 113 The low cell-based activity of 5,6-dihydro-AZA can be attributed to its inefficient conversion to the activated deoxyribonucleoside triphosphate, leading to poor DNA incorporation. 139 5-Fluoro-2′-deoxycytidine (5F-dC), when incorporated into DNA, inhibits DNMT1 through an irreversible covalent complex similar to AZA and DAC.140,141 The cellular metabolism of 5F-CdR does not result in its conversion to 5F-dCTP or subsequent DNA incorporation, instead being converted to 5-fluoro-2′-deoxyuridylate, an inhibitor of thymidylate synthetase. 142 Zebularine-incorporated DNA also inhibits DNMT1 through a covalent, though reversible, complex. 141 Similar to 5,6-dihydro-Aza-C, high concentrations of zebularine (~50 µM) are required to observe DNMT1 inhibition in cells due to inefficient incorporation into DNA. 143
Oligonucleotides
MG98, a 20-bp antisense oligonucleotide, was developed to prevent the translation of DNMT1. The sequence is complementary with the 3′-UTR of the DNMT1 mRNA and has shown DNMT inhibitory activity in a preclinical xenograft model. 144 Unfortunately, clinical trials using MG98 have failed to demonstrate activity.145–147 miR29a, a microRNA that targets DNMT1 and DNMT3a/b, has been shown to reduce DNA methylation in cells. 148
Reversible inhibitors
Despite the successes of AZA and DAC, there is significant need for DNMT inhibitors that do not rely on DNA incorporation for activity. A recent study using a panel of human cancer cell lines showed a 1000-fold variability in DAC potency, attributed to differential incorporation of DAC into DNA. 149 As DNA hypomethylation correlates with clinical benefit of DAC in MDS, 131 it is possible that some element of MDS resistance to AZA and DAC stems from the ability of tumor cells to block the import, phosphorylation, and incorporation of the drugs into DNA. Nonincorporating inhibitors could bypass these hurdles by targeting DNMT directly.
Nonincorporating DNMT inhibitors may offer a large therapeutic window, compared to the U-shaped dose–response curve for DAC and AZA limited by cytotoxicity and lack of DNA incorporation at high concentrations.116,117 In addition, the development of selective compounds for DNMT1, DNMT3a, and DNMT3b would facilitate targeting of diseases driven by a particular DNMT enzyme, with minimal off-target effects. Selective DNMT inhibitors would also have value as tool compounds to understand the biological roles of the individual DNA methyltransferases.
A number of small molecules have been reported to inhibit DNA methylation, either through in vitro biochemical or cell-based assays. Although the following list is not exhaustive, it demonstrates the diversity of compounds that were reported to inhibit DNMT activity ( Fig. 2B ).
Hydralazine is a cardiovascular drug that was shown to inhibit DNA methylation in cells,150–152 although not in an in vitro biochemical assay. 152 The DNMT inhibitory activity of hydralazine is controversial, as a subsequent study was unable to reproduce DNMT inhibitory activity in cells.153,154 Clinical studies of hydralazine in combination with a histone deacetylase inhibitor (valproate) in MDS are under way. 155
Procainamide is another cardiovascular drug with apparent activity against DNMT in cells.150,151 Like hydralazine, however, the DNA methylation inhibition of procainamide in cells has been disputed. 153 Procaine, a closely related molecule, has also been reported to inhibit DNA methylation in cells, 156 although this effect was not confirmed by an independent follow-up study. 157
(–)-Epigallocatechin-3-gallate (EGCG), a major component of green tea extracts, has been shown to inhibit DNA methylation in vitro using nuclear extracts, presumably through binding to DNMT1, as well as alterations in methylation status and reexpression of tumor suppressor genes. 158 Subsequent studies, however, failed to confirm DNMT inhibition with EGCG.153,157
RG108 is a small-molecule inhibitor of DNMT1 that was discovered using a computational screening approach. 159 The compound blocked DNA methyltransferase activity in vitro and in cells inhibited the methylation of tumor suppressor gene promoter DNA, but not DNA in centromeric satellite elements, suggesting a context-dependent DNA methylation inhibition. 160 RG108, unlike the nucleoside analogs AZA, DAC, zebularine, procaine, and EGCG, did not demonstrate cytotoxic or genotoxic effects on cells even at high concentrations. 157
The antibiotic mithramycin A was shown to reduce methylation at tumor suppressor gene CpG islands and reactivate the tumor suppressor genes in cells. In addition, treatment with the drug resulted in DNMT1 depletion as well as decreased mobility of cancer cells. 161
SGI-1027, a quinoline-based compound, has demonstrated inhibitory activity against DNMT1, DNMT3a, and DNMT3b in biochemical assays and resulted in decreased methylation at tumor suppressor gene CpG islands and corresponding gene upregulation. 162
Nanaomycin A is an antibiotic that was recently reported to selectively inhibit DNMT3B, dramatically inhibit cytosine methylation in cells, and result in the reexpression of RASSF1A, a tumor suppressor gene. 163 Other DNMT–isoform selective compounds have also been recently described, including DNMT3B-selective NSC 14778 and DNMT1-selective NSC 106084. 164
At first glance, there appear to be few underlying similarities between the various published DNMT inhibitors. As efforts to directly target DNMT enzymes mature, there will be a need for rigorous biochemical and biophysical characterization of DNMT inhibitors that is, as yet, lacking for the compounds discussed above. We have found that compounds that bind to DNA, such as the intercalator ethidium bromide, can potently inhibit DNMT enzymatic assays (unpublished data), highlighting the need for careful characterization of screening hits. However, the potential clinical benefits of reversible DNMT inhibitors justify any required validation efforts.
Challenges and Opportunities for DNMT Therapy Development
The discovery and development of novel pharmaceuticals targeting DNMTs faces significant challenges. These include but are not limited to identification of inhibitors that do not incorporate into or bind to DNA, effects of competing with SAM binding or other cofactors, how specific DNMT inhibitors will perform in the context of the cell cycle dependency of DNMT function, development of resistance mechanisms in tumors, and whether global changes in methylation will cause genomic instability, allelic imbalances, and retrotransposon reactivation. In addition, optimal pharmaceutical properties of novel inhibitors will be vital given the potential for toxicities associated with DNMT inhibition and whether drug stability issues or confounding metabolite generation will affect maintenance of methylation patterns and concomitant gene reexpression in tumors versus normal tissues. Despite these challenges and given the success of marketed hypomethylating drugs, targeting DNMTs remains an attractive approach for the development of novel cancer therapies.
The approach of directly inhibiting DNMTs with nonnucleoside inhibitors offers an opportunity to selectively inhibit different members of the DNMT family of enzymes. The X-ray crystal structure of the human DNMT3a/3L complex was first published in 2007, and more recently, the X-ray structures of both human and mouse DNMT1 were described.43,165 However, no DNMT structures in complex with small-molecule inhibitors, other than with the cofactor reaction product S-adenosyl-homocysteine (SAH), have been reported. These structures should assist in the screening, discovery, and development of novel inhibitors targeting DNMTs. A list of small-molecule DNMT inhibitors discovered to date, including those described above ( Fig. 2 ), can be found in public databases (ChEMBL and The Binding Database).166,167
Many inhibitors with a structure–activity relationship (SAR) around blockade of DNMT1 or DNMT3b activity have been described and offer the opportunity to explore refined effects on the hypomethylation of the genome through the blockade of de novo methylation, maintenance methylation, or both.168,169 The potential advantage of separate selective inhibitors for DNMT1 and DNMT3b clinically would enable the titration of drugs to these targets depending on the tumor type and potentially balance any toxicity that each drug may engender. Targeting of DNMT3a may provide alternative hypomethylating effects, but two recent reports indicate that DNMT3a mutations that may be functionally inactivating occur in AML, suggesting that blockade of that enzyme could promote tumor formation after long-term inhibitor treatment.50,51 Additional effective inhibitors may be those that concomitantly inhibit DNMT1 and DNMT3 enzymes and could have efficacy similar to the nucleoside analogs. A nonnucleoside pan-DNMT inhibitor may phenocopy the effects observed by AZA or DAC without incorporation into DNA, thereby reducing the potential for chromosomal instability and bone marrow toxicity related to DNA damage. Avoidance of dose-limiting myelotoxicity through direct DNMT inhibition may support combination treatments with chemotherapeutic agents. Induced chromatin remodeling may lead to secondary tumors due to modulation of DNA methylation patterns as observed in DNMT1 genetically altered mice.104,105 Ideally, novel inhibitors of DNMTs would not bind to or incorporate into DNA since intercalation or minor groove binding may lead to genomic instability and potential toxicity and would mediate their effects through competition with SAM binding and catalytic disruption. Studies have demonstrated that normal cells are relatively resistant to AZA and DAC treatment, in part due to their reduced proliferative capacity.110,111 Direct inhibition of DNMTs with nonnucleoside drugs should correspondingly have limited toxicity in normal tissues.
Understanding how individual nonnucleoside inhibitors of DNMT1, DNMT3, or combination drugs specifically affect promoter methylation, gene expression, and tumor growth will be critical for clinical assessment of efficacy. Knowing which CpG sites are affected by hypomethylation through blockade of the different enzymes, as well as to what degree demethylation results in transcriptional modulation of specific functional genes, will facilitate biomarker development. Another challenge involves whether reversible or covalent inhibitors should be developed and how these different approaches will affect the clinical therapeutic window. These challenges await the discovery of broader genomic methylation and expression patterns in tumors, combined with innovation in fragment-based chemistry approaches and docking-based virtual screening of enzyme active sites. 169
Another complicating factor in the development of DNMT inhibitors is the contribution of the nucleosome and its associated histone marks in the expression of genes.14,170 The reexpression of genes following hypomethylation of DNA requires the eviction of nucleosomes from gene promoters. Treatment of cells with DAC led to demethylation of CpG islands, but not all of them had a corresponding loss of nucleosomes upstream of transcription start sites. 171 Unmethylated promoters with intact nucleosomes were repressed despite DNA hypomethylation, possibly due to Polycomb repressor complexes that silence gene transcription through changes in specific histone marks known collectively as PRC reprogramming.14,170,172 Inhibitors of DNMT1, DNMT3, or both may lead to promoter and gene body hypomethylation but may not in all cases lead to reexpression of tumor suppressor genes, miRNAs, and other functional genes important in tumor biology. The plasticity of CpG methylation within promoters and gene bodies is both heritable and stochastic, and inhibitors will likely have effects on individual CpG sites differently. Thus, therapeutic combinations of specific DNMT inhibitors together with drugs that target histone modifications may offer an approach to address the tumor epigenome as a whole. Alternatively, targeting enzymes such as the histone methyltransferase SET7/9 or the histone demethylase LSD1 that regulate DNMT1 stability may result in a pronounced epigenetic effect.173,174
Recent examples of combination epigenetic targeting in clinical trials, including evaluation of AZA or DAC with histone deacetylase (HDAC) inhibitors, have been reviewed previously.168,175 Overall, there have been limited clinical synergies from these combinations in hematological malignancies or solid tumors, possibly due to dosing schedules, selection of the most appropriate cancer indications, or stage of disease. A better understanding of both global and specific hypomethylation and acetylation patterns induced by treatments with DNMT + HDAC inhibitors compared to the clinical response is warranted. Since HDAC inhibitors affect acetylation of nuclear histones, cytoplasmic p53, and other targets, the effects of these drugs are complex and may modulate both tumor cell survival and differentiation pathways. Additional clinical studies will help to identify when epigenetic drug combinations will be most effective, particularly when tumor debulking approaches should be combined with antimetastatic or anticancer stem cell therapies. Combinations between SGI-110 and other drugs may offer advantages in both dosing schedule and drug deposition in tumors compared with DAC or AZA. Furthermore, profiling of tumor methylation status will help to stratify patients with respect to treatment selection and associated risks. 175
Drug combination approaches also have included epigenetic targeting together with either conventional chemotherapy or selective therapies such as Gleevec. 168 The conclusions from these trials included both improved clinical benefit and hypomethylation of specific gene promoters,176-180 opening the possibility for evaluation of yet untested combinations. Current strategies include combinations of hypomethylating agents with biologics, including Vectibix, Rituxan, Avastin, or pegylated-IFN-α2b, as well as small molecules such as Velcade or Nexavar (clinicaltrials.gov). As the development of new inhibitors of specific DNMTs and other epigenetic targets emerges, 181 the opportunities for testing combinations of epigenetic drugs together or combined with appropriate targeted therapies that approach different signaling pathways in tumors will expand significantly. 182 Epigenetic drug development will continue to enhance the armamentarium of therapies useful in the treatment of a variety of malignancies, which will ultimately address the significant unmet need remaining in the field.
Footnotes
Acknowledgements
The authors thank Jeremy Lamb for his instrumental help with the figures and Stuart Hwang for suggestions and critical review.