
Abstract
Epigenetic control of the transciptome is a complex and highly coordinated cellular process. One critical mechanism involves DNA methylation, mediated by distinct but related DNA methyltransferases (DNMTs). Although several DNMT inhibitors are available, most are nonselective; selective DNMT inhibitors, therefore, could be optimal as therapeutics, as well acting as chemical probes to elucidate the fundamental biology of individual DNMTs. DNA methylation is a stable chemical modification, yet posttranslational modification of histones is transitory, with reversible effects on gene expression. Histone posttranslational modifications influence access of transcription factors to DNA target sites to affect gene activity. Histones are regulated by several enzymes, including acetylases (HATs), deacetylases (HDACs), methyltransferases (HMTs), and demethylases (HDMTs). Generally, HATs activate, whereas HDACs suppress gene activity. Specifically, HMTs and HDMTs can either activate or inhibit gene expression, depending on the site and extent of the methylation pattern. There is growing interest in drugs that target enzymes involved in epigenetic control. Currently, a range of high-throughput screening (HTS) technologies are used to identify selective compounds against these enzymes. This review focuses on the rationale for drug development of these enzymes, as well the utility of HTS methods used in identifying and optimizing novel selective compounds that modulate epigenetic control of the human transcriptome.
Introduction
Epigenetics refers to the control of gene expression by factors and mechanisms beyond the DNA sequence itself. 1 Specifically, direct chemical modification of DNA, such as occurs during DNA methylation, can block the binding of transcription factors to DNA to inhibit gene expression.1–3 Similarly, posttranslational modifications of histones can also affect DNA conformation, as well as DNA interaction with transcription factors. 4
The physiological control conferred by epigenetics produces fundamental changes in cell activity. Although most cells possess the same genotype, they almost all have different phenotypes. This is primarily due to epigenetic control generating different patterns of gene expression, resulting in different proteins expressed, which discretely alters changes in the makeup of each cell and their functions. Consequently, epigenetic control mechanisms are critical in organismal development and differentiation to distinct cell lineages and phenotype. 5 Two of the major forms of epigenetic control mechanisms are DNA methylation and histone modification.
DNA Methylation
DNA methylation is catalyzed by a family of DNA methyltransferases (DMNTs). 1 The reaction involves the transfer of a methyl group from S-adenosyl methionine to the 5′ position of a cytosine ring of CpG dinucleotides. CpG dinucleotides tend to cluster in regions referred to as CpG islands.1–3 Although CpG dinucleotides are relatively uncommon in the genome, they tend to cluster near promoter regions and therefore exert a disproportionate influence on gene expression and activity. More than half of all promoters in the human genome have CpG islands and therefore are subject to control by DMNTs. 1 However, in most normal cells, the CpG islands are not methylated, and the genes remain active.
Extensive studies aimed at identifying the methylomes of cells at distinct states of differentiation have led to a better understanding of the role of epigenetics in organismal development. Consequently, DNA methylation has also attracted considerable interest in the field of stem cell development and in the generation of induced pluripotent stem cells, where differentiated states of the cells must be reprogrammed, and this involves modulation of DNA methylation. 5 Furthermore, DNA methylation is involved in inactivation of the X chromosome, as well as repetitive elements, and in the control of imprinted genes and tissue-specific genes.1,6
Abnormal DNA methylation plays a role in the pathophysiology of autoimmune diseases,7–10 and there is likely to be a role of DNA methylation in numerous diseases, yet perhaps the most studied is its role in cancer.2,3 This is because hypermethylation, occurring at CpG islands, causes inhibition of tumor suppressor. Because the methylation reaction is stable, DNA methylation patterns or “fingerprints” have now been identified for a range of cancers with attempts being made to employ these patterns as biomarkers for cancer classification. Ordway et al. 11 reported that in a genome-wide analysis of DNA methylation patterns of breast cancer samples, an array of methylated sites was detected that were distinct from the DNA of nonmalignant tissue. They also showed that methylation of one specific gene showed specificity with regard to normal breast tissue of nonmalignant breast cancer. From these studies, they were able to develop a panel of biomarkers that could be used as breast cancer diagnostics.
Fang et al. 12 employed DNA methylation analysis of breast cancer tissue to identify potential biomarkers of the metastatic nature and aggressiveness of the disease. The methylation patterns were closely associated with genes in the so-called metastasis transcriptome. Hypermethylation of this region was associated with lower metastasis and increased patient survival, whereas hypomethylation was associated with a greater risk of disease progression. Consequently, methylome analysis should provide an approach to identify cancers that need to be more aggressively treated because of their increased potential to spread and systemically proliferate.
In a similar context, Rauch et al. 13 identified methylation fingerprints in lung squamous cell carcinomas as potential lung cancer biomarkers. Similarly, Dunwell et al. 14 conducted a genome-wide analysis of childhood acute lymphoblastic leukemia and not only identified methylation patterns that distinguished cancerous from noncancerous cells but also distinguished malignant B from T lymphocytes. This hypermethylation was also associated with a diminished efficacy of treatment by the drug imatinib. Consequently, methylation fingerprints can provide insights into disease diagnosis and could be useful in identifying the most appropriate kind of treatment regimens.
Collectively, the methylation of several genes can be used as a predictor of efficacy for particular drug treatments with such patient profiling being used to prioritize therapeutic options. As reviewed by Kelley et al., 2 the lack of methylation of the promoter regions of some genes may indicate responsiveness to cisplatin chemotherapy. Conversely, treatment with Food and Drug Administration (FDA)–approved DNMT inhibitors increases expression of some genes that enhance sensitivity of some cancers to cisplatin therapy, suggesting that DNMT inhibitors can increase efficacy of established chemotherapeutics. Methylation patterns have also been linked to efficacy of tamoxifen treatment of breast cancer and bladder cancer to interleukin (IL)–2 therapy. DNMT inhibitors have also been found to increase sensitivity of metastatic melanoma cancers to chemotherapy.
Methylation Inhibitors and Drug Discovery (see Table 1)
Modulation of epigenetic control systems holds promise in the development of novel drugs and therapeutics. Drugs that inhibit DNMTs have some potential advantages as chemotherapeutics, particularly as they act primarily during the S phase of the cell cycle and thus affect rapidly proliferating cells. Two DNMT inhibitors that have been approved by the FDA to treat the myeloid cancers, cutaneous T cell lymphoma, or myelodysplastic syndrome (MDS) are 5-azacytidine (5-Aza-CR; azacitidine [Vidaza]) and 5-aza-20-deoxycitidine (5-Aza-CdR; decitabine [Dacogen]).14–16 Both are cytosine analogs that act by incorporating into DNA and covalently binding DNMTs via their modified pyrimidine resides. They reduce levels of the DNMTs by targeting the enzymes for proteosomal degradation. Both possess significant toxicity at high concentrations due to DNA damage. More potent and selective DNMT inhibitors potentially with less toxicity are in development, including zebularine.17,18
DNA Methylation Enzymes

Due to their abilities to produce global demethylation, both azacitidine and decitabine have been clinically tested with other FDA-approved chemotherapeutics in clinical cancer trials. Indeed, both agents, at low doses, appear to enhance efficacy of lenalidomide, carboplatin, imatinib, and gemtuzumab for the treatment of several cancer types.3,15,18 These studies support the utility of DMNT inhibitors both as single-drug cancer therapeutics and, more important, in potentiating efficacy of more standard therapies in combination therapies.
Nonetheless, development of drugs that are more selective in inhibiting specific DNMTs would be useful in reducing potential toxicity while increasing efficacy. There are, however, hurdles to overcome in developing more conventional competitive and highly selective DNMT inhibitors. First, the activity of DNMT involves protein–DNA interactions, which are notoriously difficult to design small-molecule inhibitors. Second, the reaction is unusual, in that it requires the DNMT to induce the cytosine residue to “flip out” from the DNA helix. 19 However, there are important aspects of the DNMT-DNA interaction that may be useful for novel drug design.
First, there is a degree of selectivity in which CpG islands are hypermethylated in cancer cells with relatively few of the CpG islands in a DNA sequence being methylated. Although the basis for this specificity is unknown, it suggests that there are factors involved in the targeting and efficacy of DNMTs in methylating CpG sites discretely, and those factors could be used in developing selective drugs. 20 In fact, because the methlytransferase reaction involves a “flip-out” conformation of a cytosine from the DNA strand, a specific 3-D configuration of the cytosine and its associated nucleotides bound to DNMTs is likely to form. This may allow for enzyme recognition in much the same manner as protein kinases target specific substrates to cause site-specific phosphorylation.
Conceivably, structural analysis of DNA binding to the enzyme could be used to develop small-molecule inhibitors of methylation of specific CpG sites in the DNA. This would be particularly important if unique pharmacophores could be developed from the interaction of DNMT with CpGs in promoter regions of tumor suppressor genes. In fact, studies by Handa and Jeltsch 21 suggest that flanking sequences of CpG dinucleotides are critical for conferring specificity of DNMT3a- and DNMT3b-catalyzed methylation. They identified flanking sequences of four nucleotides around CpG that were associated with greatly increased methylation, as opposed to those flanking sequences for which limited methylation occurred. They then tested experimentally whether those flanking regions actually affected methylation and showed over 13-fold differences in methylation based on the four nucleotides flanking the CpG regions. These authors concluded that flanking regions around the target methylation sites were critical for preference of DNMT3a and DNMT3b and also suggested that the flanking sequence preferences could be involved in the origin of CG islands. Thus, modeling of CpG and the preference flanking regions could provide an approach to identify pharmacophores for the interaction of DNMTs with their targets.
In general, if the flipped-out conformation of cytosine, as well as its corresponding adjacent nucleotides, forms a specific structure that the DNMT recognizes, then structure-based design could be used to discover selective disinhibitors of each gene normally regulated by DNMTs, resulting in selective gene activation. Siedlecki et al. 22 and Brueckner et al. 23 have developed a 3-D model of the catalytic domain of DNMT1, which they used for an in silico screen of small-molecule libraries. Using this approach, they identified a small molecule RG108, which they showed was a potent competitive inhibitor of DNMT1 that blocked DNA methylation. Furthermore, they reported that RG108 reversed methylation of previously silenced tumor suppressor genes in human colon cancer cells. This suggests that rational approaches based on structural analysis of the DNMTs could be employed to discover selective and potentially less toxic enzyme inhibitors.
Kuck et al. 24 extended these studies by employing a multistep structure-based virtual screening of more than 65 000 compounds to identify small molecules that selectively inhibited DNMT1 versus DNMT3. They provided the first selective inhibitors of a DNMT enzyme. This study supports the feasibility of employing rational drug design to identify competitive inhibitors with improved selectivity over previous inhibitors of DNA methylation.
DNMT Screening Assays for Drug Discovery (see Table 2)
DNMT assays
One factor that has delayed progress in developing highly selective DNA methylation inhibitors relates to existing laborious assays that measure DNMT activity. For example, Siedlecki et al. 22 and Brueckner et al. 23 used assays in which the readout was based on the ability of a methylation-sensitive restriction enzyme to cut a PCR fragment. If the DNA was methylated, the enzyme cleaved the DNA and the fragment was detected by gel electrophoresis.
Assays for DNMTs, HATs, and HDACs
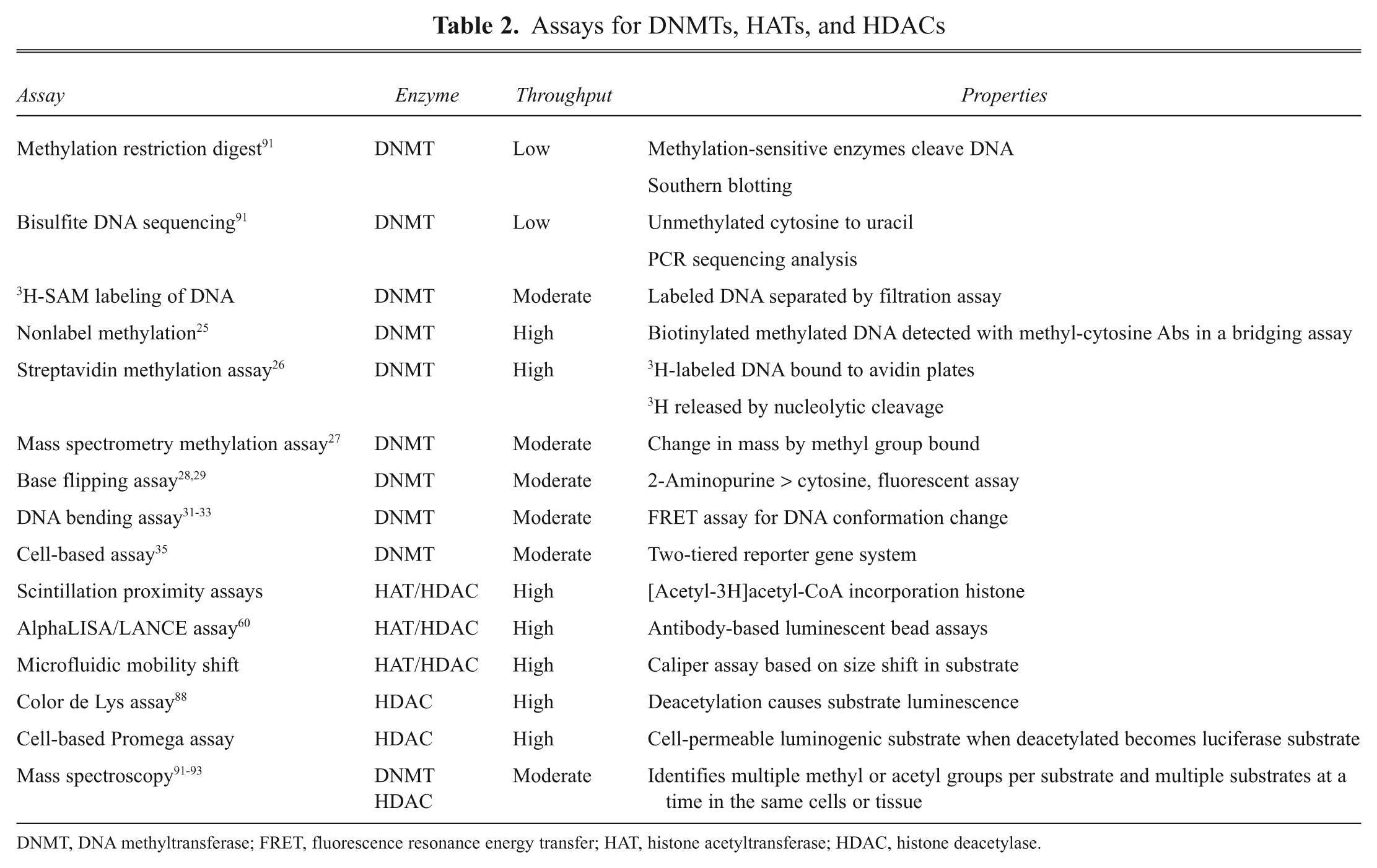
DNMT, DNA methyltransferase; FRET, fluorescence resonance energy transfer; HAT, histone acetyltransferase; HDAC, histone deacetylase.
A less cumbersome assay was used by Kuck et al. 24 in testing compounds derived from their virtual screen. Here, PCR fragments with known CpG methylation sites were incubated with recombinant DNMT1 or DNMT3 in the presence of 3 H-SAM ([methyl- 3 H] AdoMet; PerkinElmer, Waltham, MA). The assay measured the incorporation of 3 H-methyl groups into the DNA, which was then separated from the reactants in a subsequent filtration assay.
A nonisotopic high-throughput screening (HTS) assay has been developed by Rouleau et al. 25 to identify inhibitors of DNMT. In the assay, biotinylated oligonucleotides containing target CpG methylation sites are reacted with SAM and recombinant DNMT. The incorporation of the methyl group into the cytosine is detected in a homogeneous bridging assay. The assay format has been set up for the three major DNMTs (DNMT1, 3a, and 3b), shows high signal to noise (40-fold), and requires little DNMT (10 nM) with intra-assay variation of 7%. Furthermore, the assay was sensitive to known DNMT inhibitors. Consequently, the assay could be employed for HTS of compounds that selectively inhibit the different DNMTs.
Other assays that have been used to measure DNMT activity involve a streptavidin assay format in which the 3 H-methylation reaction of DNMT with an oligonucleotide occurs in solution, the labeled DNA is bound to avidin-coated microplates, and radioactivity incorporated into the DNA is released by a nucleolytic digestion of the DNA. 26 This assay has high sensitivity, low background, and good accuracy and reliability, and it is easily adapted for HTS formats. Furthermore, Humeny et al. 27 developed a nonradioactive matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) assay that measures methyl group incorporation by change of mass in an oligonucleotide substrate that could also be employed for HTS compound screening. The assay provides the advantage of being able to measure methylation of several different sites simultaneously.
Novel DNMT assays
Base flipping assays
An unusual action of DNMTs in methylating CpGs occurs when the enzymes bind to DNA and induce conformational changes, resulting in the flipping of cytosine around the phosphate backbone, away from the DNA helix and into the reactive site of the DNMT. Base flipping is essential for the DNA helix to move to an open conformation, allowing DNMT to transfer methyl groups to the normally hidden cytosines in the chromosome. Assays using base flipping can be used to identify DNMT inhibitors.
One approach in this regard involves substitution of the fluorescent analogue of adenine, 2-aminopurine (AP), for cytosine in target DNA. AP fluorescence is highly quenched within the structure of double-stranded DNA but enhanced if opened to the solution. Such perturbations of the duplex DNA structure are induced by DNMT binding, and the binding reaction resulting in base flipping out of the DNA helix can be monitored as a change in AP emission intensity. 28 Importantly, AP fluorescence excitation and emission maxima are discretely separated from those of tryptophans and tyrosines in proteins surrounding the DNA, increasing the usefulness of the approach for studying protein–DNA interactions. Furthermore, Neely et al. 29 developed a time-resolved fluorescence assay to measure the fluorescence decay function of AP with respect to base flipping, providing greater specificity of the flipping assay and for DNMT binding. When AP is not localized to its specific target site, appropriate fluorescence decay is not seen. Taken together, the approach not only provides for detection of binding of DNMT to its selective target sites but also indicates that the resulting conformational change is directly associated with “flipping” rather than other potential conformational changes arising from non-DNA methylation reactions.
In addition to base flipping assays, several other approaches measure the interaction of DNMT with target DNA sites as potential assays to identify novel and selective DNA methylation inhibitors. Prior to DNMT-induced base flipping, the molecule targets specific binding sites on DNA, inducing bending of the DNA strands. It has been estimated that DNMT induces between a 50° and 60° bend in the DNA, which appears to be part of the ability of the enzymes to unravel the DNA helix, allowing cytosine access to the DNMT active site.
Studies with model DNMTs have shown that the enzymes consist of at least two domains: a catalytic domain involved in methyl transfer to DNA, similar among most DNMTs, and a DNA binding domain. The DNA binding domain creates specificity in the sites of DNA modified by DNMTs. This is important because most CpG dinucleotides in the genome are unmethylated, and therefore the DNA binding domain for DNMTs selects for those methylated sites. Mutations in only a few of the amino acid residues of the DNA binding domains of DNMTs significantly affect DNA targeting of the enzymes.30,31 Furthermore, chimeras of the catalytic domains of DNMTs fused to DNA recognition sites of transcription factors can target hypermethylation of selected CpGs and promoters, indicating that the DNA binding domains are critical in targeting DNMT function.
The DNA “bending” reaction induced by DNMTs can be detected using fluorescence resonance energy transfer (FRET)–based assays. For these assays, oligonucleotides corresponding to regions involved in DNMT methylation are tagged with flurophores. The bending reaction evokes a change in fluorescence emission.31,32 In general, however, these assays are both cumbersome and to date have mainly been employed in basic research studies, as opposed to compound screening. However, recent reports by Ricci et al. 33 have developed an electrochemical format for measuring protein–DNA binding that could be employed to discover compounds that inhibit binding of DNMTs to selective sites in the genome. These workers reported a chemical sensor consisting of oligonucleotides with redox-tags covalently bound to electrodes. When a protein binds to the DNA, current flowing through the oligonucleotide is reduced, thereby providing a highly sensitive, label-free assay of protein–DNA binding. They showed that this approach could be used to measure binding of DNMTs to target DNA. It was sensitive at nanomolar protein concentrations, was specific for protein binding sites, and provided rapid responses within minutes. Given these characteristics, it is possible that the technology may be adaptable for HTS.
This group has also developed a total internal reflectance fluorescence (TIRF) spectroscopy assay that can also measure DNMT binding as well as other epigenetic factors, including histone acetyltransferases (HATs) and histone deacetylases (HDACs). 34 The technology employs hydrogel double-stranded DNA microarrays and dye-labeled regulatory proteins that detect multiple protein–DNA interactions in a kinetic manner. In the microarray format, arrays are generated using hydrogel spots containing different double-stranded DNA fragments. Microfluidics are used to introduce sample and reactants to the spots and employ hydrogels to overcome the artifacts associated with DNA attachment to solid surfaces. The array acts as a waveguide to generate TIRF in the flow cell chamber that optically measures binding of fluorescently labeled proteins to DNA. The study demonstrated that the technology could be employed to measure binding of transcription factors to different sequences of DNA. In addition, the binding intensity varied, with DNAs containing cognate binding sites for transcription factors. In essence, the intensity of binding reflected those proteins bound to their predicted binding sites in the DNA. Consequently, the technology may be adaptable for a range of nuclear binding protein and thus amenable for small-molecule screening.
Cell-based DNA methylation assays
To complement in vitro DNMT assays, Lin et al. 35 have developed a cell-based assay to measure DNA methylation by employing a two-tiered reporter gene system. Specifically, constructs for the Tet repressor under the regulation of the Trip10 promoter were generated. The Trip10 promoter possesses multiple CpG sites for DNA methylation such that when the constructs are methylated, the Tet repressor is turned off. This allows the Tet operator on a separate construct to generate enhanced green fluorescent protein (EGFP) and thus generate a fluorescent signal. Thus, DNMT inhibitors will cause a reduced green fluorescent protein (GFP) fluorescent response in the cell. The assay format was validated against a small-molecule library, and several novel DNMT inhibitors were identified. Conceivably, this assay, together with high-throughput in vitro assays, could be employed to identify novel and selective inhibitors of the different DNMTs, leading to novel drugs acting to block DNA methylation.
Histone Methylation (see Table 3)
In addition to DNA methylation, epigenetic control of gene expression can be modulated by methylation of histones at lysine and arginine residues, and this is catalyzed by a family of enzymes distinct from those involved in DNA methylation.1,2,4,36,37 Methyl groups can be added to lysine residues at three different sites as either mono-, di-, or trimethylations and two different sites on arginine residues. 37 The number of methylations and the specific sites methylated on a given residue can produce different effects on chromatin structure and gene expression.37,38
Histone Methylation and Demethylation Enzymes
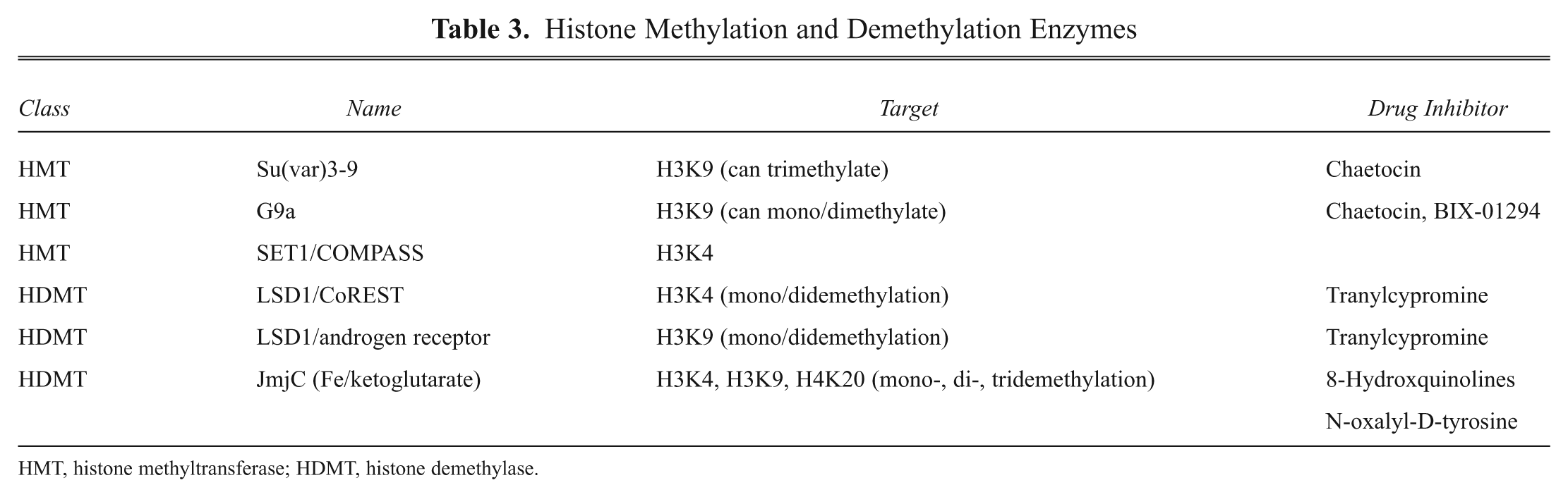
HMT, histone methyltransferase; HDMT, histone demethylase.
The two major sets of enzymes involved in histone methylation are histone methyltransferases (HMTs) and histone de- methylases (HDMTs). The HMTs can be further subdivided into those enzymes involved in methylating lysine residues (HKMTs) and those that methylate arginines (HRMTs). 4
The most extensively studied histone methylation event involves methylation at lysine 9 on histone, 3 referred to as histone 3 lysine 9 (H3K9).2,4,37,39 Other important histone methylations are those at H3K4, H3K27, H3K36, H3K79, and H4K20. Those methylation events most closely associated with actively transcribing genes are H3K4, H3K36, and H3K79, whereas methylation of H3K9 and H3K27 is generally linked to gene silencing and chromatin condensation.1,4
Aberrant histone methylation has been linked to disease, most prominently cancer.2,4,39 In fact, some efforts have been made to link histone methylation to cancer disease subtyping. For example, the pattern of H3K4 methylation has been associated with occurrence of prostate cancer, whereas demethylation of H3K9 has been suggested to distinguish cancerous from noncancerous prostate tissue. Furthermore, the overexpression of the HRMT, CARM1, has been associated with a selective stage of breast and hormone-dependent prostate cancer. 4
Histone Methylases
One of the first HMTs identified, Su(var)3-9, selectively methylates H3K9, which leads to gene silencing.36,37 Consequently, drugs that selectively block Su(var)3-9 would be expected to increase gene expression. Su(var)3-9 commonly works together with another HKMT, G9a, to methylate H3K9. G9a appears to primarily mono- and dimethylate H3K9, whereas Su(var)3-9 is involved in di- and trimethylation of this residue.40–43 A third HKMT, SETDB1, can produce all three states of methylation of H3K9. The coordination of these overlapping enzymes in methylating this critical residue in epigenetic silencing appears to involve unique targeting mechanisms, including DNA binding partners, small RNAs, and DNMTs.
Even more complex is the mechanisms governing H3K4 methylation. This residue is critical for gene activation. The first HKMTs found to selectively methylate H3K4 consisted of the methylase Set1 and the macromolecular complex COMPASS, a family of conserved proteins.44,45 The composition of the COMPASS determines whether H3K4 is mono-, di-, or trimethylated and may also contribute to tissue selectivity and developmental regulation of the methylase activity. In fact, seven COMPASS-like complexes regulate H3K4 methylation. Some of these complexes involve Set1 and another family of methyltransferases, referred to as the mixed-lineage leukemia (MLL) gene.45,46 MLLs are involved in a number of human acute leukemias, and at least four different MMLs have HKMT activity and can complex with Set1 to regulate H3K4 methylation. 46 These findings provide direct links between H3K4 methylation and proliferative disorders and provide a strong rationale for the development of Set1 and MML1 inhibitors as anticancer drugs.
Histone Demethylases
As in the case of the DNA methylases, histone methylation is balanced by a family of demethylating enzymes. There are two major families of demethylases: those that remove mono- and dimethylations and those that remove all methyl groups from histone lysines. The former class of enzymes is typified by lysine-specific demethylase 1 (LSD1).2,36,39,47 LSD1 is a monoamine oxidase that oxidizes the amine group of a methylated lysine to form a formaldehyde and the corresponding amine. 36 It requires flavin adenine dinucleotide (FAD) as a cofactor. LSD1 can only demethylate mono- and dimethylated lysines because of its requirement to act upon a protonated methyl-ammonium group.
Importantly, the selectivity of LSD1 with regard to which lysines in histones to demethylated is dependent on the nature of its binding partner complex. Thus, when LSD1 associates with CoREST, it primarily demethylates H3K4.36,39 When it associates with the androgen receptor, LSD1 demethylates H3K9. 2 Thus, depending on which partner it associates with, LSD1 activity can result in activation (demethylating H3K9 to allow androgen receptor activity) or inhibition (demethylating H3K4) of gene expression.
In fact, the interaction with transcription factors may be one mechanism by which LSD1 and other HDMT are targeted to specific histones and genes. In addition to the androgen receptor, LSD1 physically associates with the estrogen receptor and is involved in estrogen-mediated gene activation. 39 Furthermore, LSD1 also associates with other histone-regulating enzymes, particularly HDACs. In fact, one way LSD1 demethylates H3K4 is by associating with HDAC1/2, which first deacetylates histone 3 proteins, allowing binding of CoREST to then recruit LSD1 to demethyalte H3K4. 38 This is a common theme of most epigenetic regulatory enzymes: Rather than acting in isolation, they generally work in concert to modulate histone proteins, providing a high degree of specificity of post-translational modification (PTM). In fact, LSD1 can also demethylate DNMT1, 2 to indirectly affect DNA methylation.
Interestingly, the crystal structure of LSD1 bound to a peptide inhibitor has been resolved and shows that the active site appears to be preformed—that is, its structure does not change upon substrate binding.48–50 Consequently, the abilities of LSD1 to demethylate H3K4 versus H3K9 seem to be very dependent on variations in partner protein interactions to target the enzyme to distinct sites on histones to regulate.
In contrast to LSD1, a different family of enzymes containing the Jumonji-C (JmjC) motif is able to demethylate lysines in histones with three methyl groups as well as demethylating mono- and dimethylated lysines because they do not require a protonated nitrogen in the substrate for activity. 39 These enzymes are the largest family of HDMTs and uniquely require alpha-ketoglutarate and iron for activity. There are at least 30 JmjC demethylases in the human genome, each having unique target specificities.39,51
Some of the specificity of action of the JmjC HDMTs is due to unique targeting sequences in the enzyme. For example, the plant homeodomain (PHD) of JMJD2A is critical for targeting the enzyme to trimethylated H3K4, H3K9, and H4K20.52,53 These domains have been found in other enzymes targeted to chromatin, and structural studies have revealed that these domains allow the JmjC HDMTs to interact with the chromatin structure to orientate the catalytic site of the enzymes with the appropriate target residues. Furthermore, the JmjC HDMTs do not work alone in demethylating histones, and members have been found to work in coordination with LSD1 and the androgen receptor to demethylate H3K9. 54 This association may explain studies linking JmjC HDMTs to androgen-dependent prostate cancer.
Histone Methylation–Based Drugs
To identify selective inhibitors for Su(var)3-9 and G9a that block H3K9 methylation, Greiner et al. 55 screened libraries of natural products to identify the fungal toxin chaetocin that blocks H3K9 methylation by inhibiting Su(var)3-9, G9a, and other H3K9 methyltransferases with low micromolar potencies. In follow-up studies, Kubicek et al. 56 employed HTS assays to screen small-molecule libraries to identify BIX-01294, a highly selective inhibitor of G9a. This compound inhibited H3K9 mono- and dimethylation at specific targets in cell-based assays. This small molecule competes with H3K9 lysine in the active site of G9a to inhibit activity. This was based on crystal structure analysis of BIX-01294 binding to G9a. 57 Such analysis is likely to provide further insights into identifying novel selective inhibitors of G9a with higher potency and more desirable pharmaceutical properties.
In addition to the HMT, major efforts have been made to identify selective inhibitors of histone demethylases. In particular, initial efforts were made to identify selective inhibitors of LSD1 because this enzyme demethylates H3K4, a site on histones involved in activation of tumor suppressor genes that are turned off in some human cancers.
Interestingly, LSD1 has monoamine oxidase activity and appears to function as a demethylase by attracting electrons from the nitrogen of the lysine in H3K4 to catalyze cleavage of the methyl groups to generate formaldehyde. LSD1 has structural similarity to monoamine oxidases A and B, which are involved in neurotransmitter and hormone metabolism. In fact, Binda et al. 58 reported that LSD1 and monoamine oxidases (MAOs) have common evolutionary origins, despite having different substrate specificities and functions. As such, monoamine oxidase inhibitors such as tranylcypromine, which are used clinically as antidepressants, were found to inhibit LSD1. Structural studies have shown that tranylcypromine acts as a suicide inhibitor of LSD1, essentially causing covalent inactivation of the enzyme. Importantly, the structural analysis on tranylcypromine binding to LSD1, including crystallography, may provide the basis for the discovery of more selective LSD1 inhibitors, which could find use as anticancer drugs. In fact, Huang et al. 47 used the similarity of LSD1 to another family of polyamine oxidases, the sperimine oxidases, to identify a unique series of guanidine derivatives that effectively inhibited LSD1 and, in a cell-based assay, not only inhibited LSD1 activity but also reexpressed previously silenced genes, suggesting that this class of compounds may have utility as anticancer agents by turning on tumor suppressor genes in cancer cells.
In addition to LSD1, major efforts have also been made to develop selective inhibitors of the JmjC HDMTs because of their prominent role in regulating trimethylated H3K4 and H3K9. Initial focus has been on developing small-molecule mimics of 2-oxyglutarate to target the ketoglutarate dependency of these enzymes for activity. To do this, King et al. 51 screened a 236 000–compound library for JMJD2 inhibitors using a fluorometric assay to measure formaldehyde production, as a measure of the demethylating activity of this HDMT, as well as mass spectroscopy to detect the loss of methyl groups from substrate. From the screening, they identified a series of 8-hydroxyquinolines that effectively inhibited activity in both cell-free and cell-based assays. Crystallographic analysis showed that these compounds mechanistically act by blocking binding to the active site iron to prevent H3K9 demethylation. This provides insights into the rational design of novel inhibitors targeting this region of the enzyme.
Interesting, in parallel studies, Rose et al. 59 used a combination of nondenaturing mass spectroscopy, combinatorial chemistry, and crystallography to identify N-oxalyl-D-tyrosine derivatives that effectively and selectively inhibited this JMJD2. Crystallographic analysis indicated that these compounds bound to the PHD domain of JMJD2, which is critical for targeting the HDMT to trimethylated H3K4, H3K9, and H4K20. Thus, these studies suggest it may be possible to discover compounds targeting unique domains of the JmjC class of HDMTs to provide selectivity with regard to the enzymes targeted and may also be useful in the design of compounds targeting selective functions of an individual HDMT. In fact, the unique aspects of both the HMT and HDMTs with regard to associating with binding partners as well as their unique structures involving binding of cofactors for catalytic activity could be exploited to identify allosteric inhibitors of the methylase and demethylase enzymes.
HMT and HDMT drug discovery assays
Many of the technologies employed to study histone methylation and demethylation are similar to those used to examine DNA methylation and, as described below, histone acetylation. For HMT, reactions can measure 3H-methyl group incorporation into histones or peptide substrates using 3 H-SAM as a donor. Alternatively, histone methylation or demethylation can be measured by immunoblotting using antibodies selective for specific methylated lysines as in the case of Huang et al., 47 who measured LSD1 demethylation of H3K4 by this approach. Similarly, mass spectroscopy can be employed to identify inhibitors of demethylases such as the G9a inhibitor BIX-01294 described by Chang et al. 57 In addition, nondenaturing electrospray ionization mass spectroscopy can be employed to detect the binding of small-molecule inhibitors to demethylating enzymes such as JMJD2, as described by Rose et al. 59 Furthermore, technologies such as RapidFire, described more extensively below in the acetylation section, can be used for HTS to measure mass differences between the methylated and nonmethylated target, whether it be a full protein substrate or peptide substrate.
The consequence of demethylation of histones is the formation of formaldehyde, and this reaction can be employed as a response to the action of demethylases on target histones or their peptide substrates. King et al. 51 described a screening method against JMJD2 to identify a series of hydroxyquinoline inhibitors. In the assay, the formation of formaldehyde is detected by its conversion of formic acid by formaldehyde dehydrogenase, resulting in the reduction of NAD+ to NADH that can be measured by fluorescence spectroscopy. The assay is easily adaptable to HTS, and the authors indicated that the Z′ is 0.85. A similar assay was employed by Rose et al. 59 in confirming the small-molecule inhibitor binding to JMJD2.
An approach that is now more commonly being used for discovery of drugs targeting methylase activity is AlphaScreen. 60 Kawamura et al. 61 adapted the technology for HTS of drugs against JMJD2. Essentially, the assay involves using a biotinylated peptide substrate of a histone protein containing a target methylated lysine. The assay is FRET based and uses an acceptor bead tagged with antibodies targeting the methylated lysine in the substrate and donor beads with streptavidin. When the lysine is methylated, the peptide substrate forms a bridge between the acceptor and donor beads, allowing for energy transfer. When substrate is demethylated, the bridge is lost, as is the FRET signal. Kawamura et al. 61 showed that the assay was homogeneous and easily amenable for HTS. Thus, this approach and those like it that employ antibodies selective for the targeted lysine or arginine methylated groups are now used extensively in drug discovery against the HMTs and HDMTs.
Histone Acetylation
Methylation is only one means of chemical modification involved in epigenetic control of gene expression. In fact, several enzymes are responsible for posttranslational modifications of histones that affect gene activity, including those that modulate acetylation.1,2,4,36 Histone acetylation is regulated by two major enzyme families: HATs (which transfer acetyl groups to histones) and HDACs (which remove the acetyl groups from histones). The interplay of these two enzyme families provides dynamic regulation of histone function. Importantly, both HATs and HDACs are targeted to active genes in the chromosome. 62
HATs transfer acetyl groups from acetyl-coenzyme A to amino groups in the lysine-rich tails of histones. Acetylation usually increases gene expression by affecting binding of transcription factors to promoter regions.1,36 It does so, in part, by reducing electrostatic interaction of negatively charged DNA with positively charged histones to open up access of the DNA to nuclear factors. Many proteins exhibit HAT activity, including p300 and the CREBP, as well as steroid receptor coactivators, SRC1, ACTR, and P/CAF.
HDACs comprise a family of enzymes that deacetylate histones.4,36,63 There are four classes of HDACs, differentiated by their sequence, structure, and cellular location (see
Histone Deacetylation Enzymes
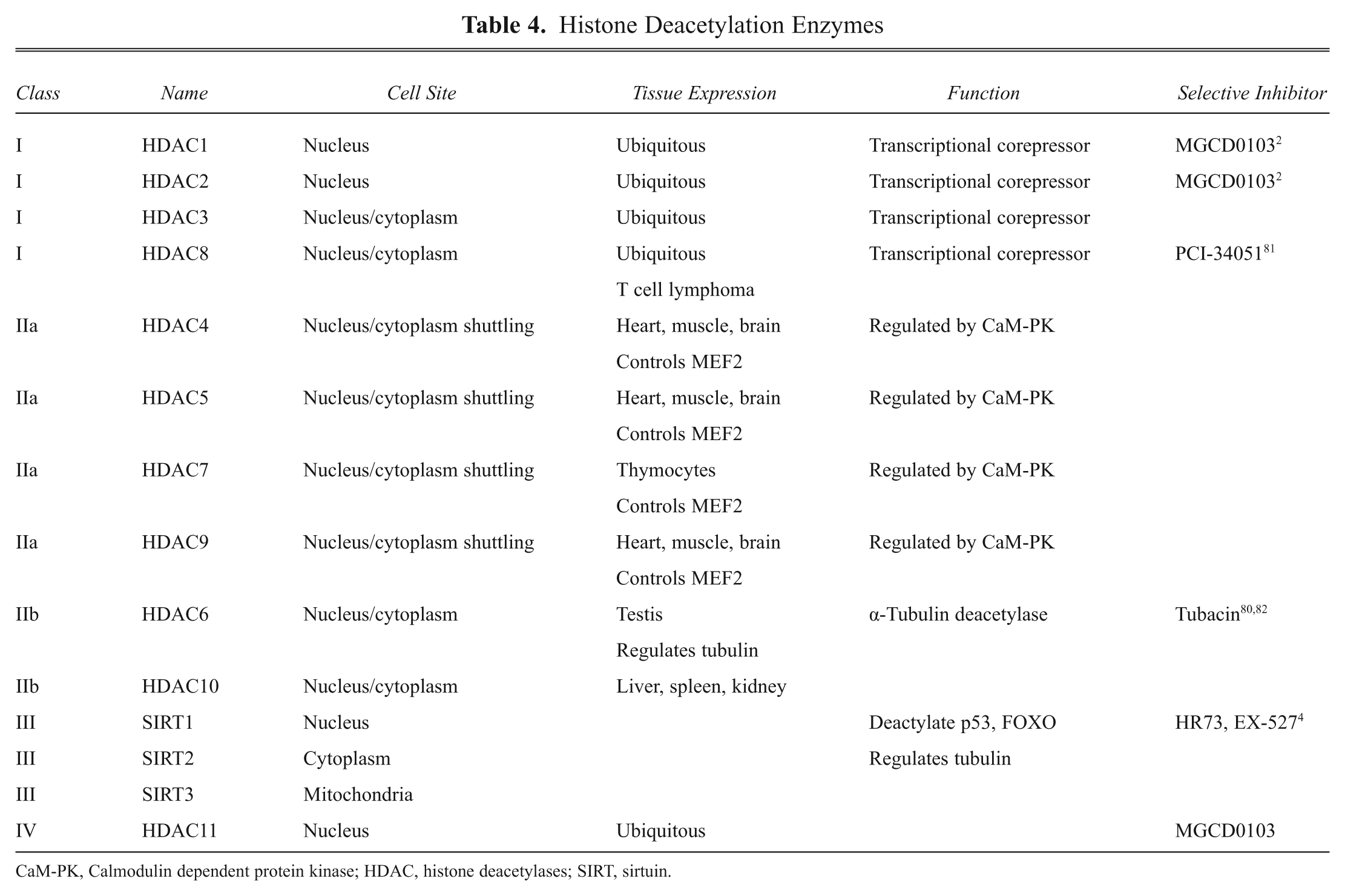
CaM-PK, Calmodulin dependent protein kinase; HDAC, histone deacetylases; SIRT, sirtuin.
For the HDAC to deacetylate histones, they need to be localized to the nucleus. They are targeted to the nucleus either via nuclear localization signals or, as in the case of HDAC1 and HDAC2, a lack of nuclear export signals. Sirtuins have both nuclear localization and export signal but are primarily expressed in the nucleus because of their association with other HDACs targeted to the nucleus. Class 1 HDACs are expressed in most cells, whereas class 2 HDACs are expressed in selective tissues such as muscle and heart. They are also found in brain, which could explain their roles in neurological diseases.
HDACs are regulated by cell signaling pathways, including kinases, such as Ca++/calmodulin kinase.64,65 The HDACs express a number of phosphorylation sites, and phosphorylation prevents their entry into the nucleus, diminishing their ability to deacetylate histones. Thus, protein kinases can influence HDAC activity by affecting HDAC translocation.
HDACs have important roles in the central nervous system in learning and memory.66,67 Neuronal activity significantly affects HDAC intracellular localization. In particular, class 2 HDACs shuttling into and out of the nucleus in hippocampal neurons are affected by spontaneous electrical activity and Ca2+ influx mediated by the activation of NMDA receptors. Sirtuins are also regulated by cellular activity. They are sensitive to NAD/NADH levels, which are affected by the redox/energetic state of the cell, due in part to mitochondrial activity. Thus, sirtuin function is also directly influenced by prevailing levels of cell activity.
Dysregulation of HDACs can cause disease, and consequently, HDACs are important targets in cancer as well as CNS and heart disease. For many cancer cells, histone acetylation is reduced, notably of the protein H4K16, due to overactive HDACs. 68 The gene silencing imparted by HDACs affects cell cycle, cell growth, differentiation, apoptosis, and angiogenesis 69 in several cancers, and there is much interest in developing HDAC inhibitors for cancer therapy.
HATs and HDACs in Disease
Most cancer cells exhibit a global reduction in histone acetylation and an overexpression of HDACs. HDACs appear to have a prime role in cancer, particularly in blood-related diseases. 70 Abnormalities in HDACs were first shown in acute promyelocytic leukemia. 70 Abnormal HDAC activity is also found in B cell lymphoma, causing transcriptional silencing. 71
Furthermore, HDACs have been implicated in a number of neurological diseases, including Huntington 72 and Alzheimer diseases. 73 In particular, SIRT1 has been shown to decline in Alzheimer disease brains and can directly deacetylate tau, 74 and overexpressing SIRT1 protects against neuronal loss in mouse models of Alzheimer disease. These findings suggest that either increasing SIRT1 activity in brain or decreasing HAT activity could be beneficial in reducing the neurodegeneration in Alzheimer disease.
HAT and HDAC Inhibitors
HAT inhibitors
Because HAT activity primarily involves transfer of an acetyl group to a lysine, the first sets of HAT inhibitors identified were peptides conjugated with acetyl-CoA. 36 Lys-CoA was found to selectively inhibit p300/CBP HAT activity. Using HTS assays, a series of isothiazolone derivatives were identified that inhibited the HAT activities of p300 and PCAF, with the compound CCT077791 being the prototypical inhibitor. 75 In efforts to identify more potent and selective HAT inhibitors, Bowers et al. 76 used a computational docking virtual screen to identify a selective p300 inhibitor, C646. This compound inhibited HAT activity in the low nanomolar range and was much more potent at p300 than other HATs. This molecule represents the first inhibitor selective for HATs, and a similar approach should be possible in developing selective inhibitors for other HAT family members.
HDAC inhibitors
Extensive pharmacological studies on HDAC inhibitors show that they generally reduce cell growth, increase differentiation, and induce apoptosis and cancer cell death. Inhibitors of HDACs cause cell cycle arrest in the G1 and G2 phases to inhibit growth, 68 and this effect is associated with the ability of the inhibitors to increase histone acetylation. Cell cycle arrest is linked to induction of p21. 77 The anticancer actions of HDAC inhibitors have also been linked to their anti-angiogenic effects, which have been linked to their abilities to reduce vascular endothelial growth factor and endothelial nitric oxide synthase expression. 78 They also produce antimetastatic effects by upregulating expression of metastatic suppressors and downregulating promoters of metastasis. 79 Like DNMT inhibitors, HDAC inhibitors can reexpress tumor suppressor genes, and HDAC and DNMT inhibitors can act synergistically to kill cancer cells.
Two HDAC inhibitors, suberoylanilide hydroxamic acid (SAHA; vorinostat) and romidepsin (depsipeptide or FK228), have been approved by the FDA to treat cancer.36,68,69 These drugs, along with other HDAC inhibitors such as depsipeptide, MS-275, and trichostatin A (TSA), have a synergistic effect in enhancing the anticancer activity of conventional anticancer drugs, including gemcitabine, paclitaxel, cisplatin, etoposide, and doxorubicin, which target malignant cells through different mechanisms. SAHA and romidepsin are nonselective HDAC inhibitors that produce global hyperacetylation. Efforts have been made to discover more selective inhibitors to avoid the side effects that occur with a global HDAC inhibitor, including cardiac and hematopoietic toxicity.
HDAC6 and HDAC8 inhibitors have been identified with a degree of selectivity.80–83 Tubacin has been shown to exhibit 70-fold selectivity for HDAC6. The HDAC8 selective inhibitor PCI-34051, which is derived from a low-molecular-weight hydroxamic acid scaffold, induces apoptosis in T cell lymphomas but not in other tumor or normal cells. 81 Other inhibitors have been identified that are selective for groups of HDAC isoforms, rather than a specific isoform, allowing their use for a wider range of diseases while minimizing side effects. 2 For example, MGCD0103, which inhibits HDACs 1, 2, and 3 (class 1) and 11 (class 4), was shown to be tolerable and to inhibit histone acetylation in patients with advanced solid tumors. MGCD0103 was also shown to be safe and to have antileukemia effects.
Some success has also been made in the discovery of sirtuin inhibitors. 4 The sirtuins are particularly important because they are involved in deacetylating H4K16, a site in histones critical in the epigenetic control of tumor suppressor genes, and sirtuin inhibitors can restore the activity of the tumor suppressors in cancer cells. HTS has identified SIRT2 inhibitors splitomycin, sirtinol, and AGK2 with low micromolar potency at blocking SIRT2 deactylating activity. Structurally different compounds have also been identified through virtual screening methods and have been reported as SIRT2 specific.83–86 A halogenated splitomicin analog (HR73) has been reported as a selective inhibitor of SIRT1. Finally, cambinol is the only sirtuin inhibitor of which the anticancer properties have been described. It was tested on Bcl6-expressing Burkitt lymphoma cells, as well as on Burkitt lymphoma mouse xenografts: In cells, cambinol induced apoptosis with concomitant hyperacetylation of Bcl6 and p53, and in the animal model, it was well tolerated and inhibited the growth of the tumors.
HAT and HDAC screening assays (see Table 2)
Traditional assays to measure HAT activity have involved use of scintillation proximity assays (SPAs) to monitor [acetyl-3H]acetyl-CoA incorporation into a histone substrate. Recently, immunoassays have been reported that selectively measure acetylated histone proteins or peptide substrates can be used. These assays measure HAT activity, as newly generated acetylated substrate, or HDAC activity, as a reduction in substrate acetylation. Several assay formats have been modified to allow for HTS of compounds. For example, AlphaLISA and LANCE assay formats (PerkinElmer) have been adapted for screening of compounds against both HATs and HDACs. 60 The AlphaLISA assay is a bead-based, chemiluminescent homogeneous proximity assay. Donor beads, coupled to streptavidin, are used to capture a biotinylated substrate. Upon illumination at 680 nm, donor beads containing a photosensitizer, phthalocyanine, then convert ambient oxygen to an excited and reactive form of singlet oxygen. In this assay, acceptor beads are conjugated to antibodies against acetylated histone H3lys9. Acetylated histone is then detected by its dual capture by the donor beads via binding of the biotin–streptavidin coupling and acceptor beads via binding of the antibody to the acetyl group bound to lysine at the 9 position of the peptide.
The LANCE assay (PerkinElmer) measures acetylation of histone H3lys9 (H3K9ac) in a time-resolved fluorescence resonance energy transfer (TR-FRET) reaction that uses a europium chelate donor dye, W1024 (Eu), together with ULight, a small molecular weight acceptor dye with a red-shifted fluorescent emission spectrum. A biotinylated histone H3-derived peptide is used as a substrate. When the substrate is acetylated, it is captured by the Eu-labeled antibody selective for acetylated H3K9 (donor) and is also bound to a streptavidin ULight-bead (acceptor). When the Eu donor and ULight acceptor dye molecules come into close proximity, the energy from the Eu donor (upon excitation at 320 or 340 nm) is transferred to the ULight acceptor dye, which, in turn, generates light at 665 nm. The intensity of the light emission is proportional to the level of biotinylated substrate modification.
Similar immunoassays have been developed to measure H3K9 acetylation using targeted antibodies to measure levels of acetylation or deacetylation. 87 Furthermore, other assay technologies have been employed for HTS of compounds against HDACs. For example, Caliper Life Sciences (Hopkinton, MA) has developed a microfluidic electrophoretic-shift format using LabChip 3000 and LabChip EZ Reader instrumentation (see www.Caliper.com). The assay uses changes in mobility shift as a measure of deacetylation of HDAC substrates, and such formats measure the deactylation reaction in real time, as well as analyzing multiple HDACs and substrates in an HTS format.
A widely used assay for HDACs is the Color de Lys assay (Biomol International, Plymouth Meeting, PA). 88 This is a colorimetric assay involving the use of a Color de Lys substrate, containing an acetylated lysine side chain. Deactylation with HDAC modifies the substrate so that reaction with a Color de Lys developer gives off an intense yellow color with absorption at 405 nm. The intensity of the yellow color is proportional to the amount of deacetylation of the substrate. This assay is simple, is homogeneous, and does not require beads or antibodies and can be used with most luminescent instrumentation. As with most colorimetric assays, however, it is prone to compound interferences.
Promega (Madison, WI) has developed a cell-based assay format to measure HDAC activity. 89 These assays generate a bioluminescent signal using a single-addition, homogeneous deacetylase format that employs a novel cell-permeable luminogenic substrate containing an acetylated lysine peptide from histone 4 linked to an aminoluciferin. This molecule is not a substrate for luciferase, but when the lysine residue is deacetylated by HDAC, the product is cleaved by a developer enzyme to provide a target for luciferase, which subsequently emits an intense luminescent signal, easily quantifiable in HTS formats.
Although these assays have become widely accepted for measuring HAT and HDAC activity, other potentially innovative approaches provide ways to identify more selective inhibitors of these enzymes. Importantly, both the HATs and the HDACs need to localize to the nucleus to be active. The enzymes undergo intracellular trafficking between the cytoplasm and nucleus and their transport can be regulated by different signaling pathways, especially protein kinases. One well-studied example of regulation of transport is that involving HDAC4, which is phosphorylated by Ca++/calmodulin kinase.64,65 The phosphorylation prevents HDAC4 localization in the nucleus, thus hindering its functions. Nuclear localization of HDAC4 can be restored if this phosphorylation is blocked. 63 Studies by Geng et al. 90 showed that the novel class 1 and 2 HDAC inhibitor, LBH589, can act by blocking translocation of HDACs from the cytoplasm to the nucleus. These studies suggest that assays to measure nuclear translocation of the acetylating and deacetylating enzymes could provide approaches for discovery of novel inhibitors, particularly when used with high-throughput confocal imaging systems.
Indeed, translocation assays have been developed as a screening approach for several molecular targets, including protein kinases and G-protein-linked receptors. They provide approaches to identify compounds that inhibit via mechanisms distinct from a blockade of catalytic sites because in many cases, the regions of the target enzymes are allosteric and distinct for the functional domains. The fact that HDACs are regulated by protein kinases at serines/threonines separate from their deacetylating domain suggests this may be the case for HDACs also. Translocation technologies are in use for drug discovery against other molecular targets (see www.DiscoverX.com). Because HAT and HDAC functions are dependent on their translocation to the nucleus, those same assay formats should be adaptable in the future for measuring HDAC and HAT translocation to allow for identification of selective inhibitors of these enzymes.
Finally, mass spectroscopy is providing a more important role in assessing histone acetylation directly, both in its use in the discovery of potential HAT or HDAC inhibitors and also in defining endogenous substrates for these enzyme in vivo.91,92 In fact, many histones that undergo acetylation can have multiple acetylation reactions at different sites within the protein that can confer different functional outcomes. Several assays described above focus on acetylation of a single target site on a peptide fragment of a histone and therefore do not provide a complete picture of the endogenous regulation of the fully intact target histone by the acetylating modifying enzyme. Mass spectroscopy, however, provides a label-free method that can easily distinguish mono- from multiple acetylations on the same full-length protein. Furthermore, HDACs may or may not act to remove all acetyl groups from a given protein, and mass spectroscopy provides an approach to determine what if any sites are still modified on the target protein. This establishes not only whether a histone is a target for a particular HDAC but on which sites on the protein the HDAC acts upon.
Interestingly, Gurard-Levin et al. 93 used peptide arrays together with mass spectroscopy to identify HDAC profiles in different cells. They generated peptide substrates for a number of different HDACs and immobilized the peptides on arrays consisting of monolayers of alkanethiolates on gold to allow for analysis of the modifications of each peptide by self-assembled monolayers for matrix-assisted laser desorption/ionization time-of-flight (SAMDI-TOF) mass spectroscopy. The arrays were then treated with nuclear extracts of different cell lines and tissues to identify HDAC activities. By measuring deacetylation, one could profile HDAC activities in each cell and provide a way to identify selective substrates for the different HDACs. Specifically, they were able to identify substrates that were selectively acetylated by recombinant HDAC2, HDAC8, and SIRT1 and determine activities of each of these enzymes on their array of substrates in different cells.
In a similar effort, Lee et al. 94 attempted to identify histone markers for the activity of HDAC1 that could be used to evaluate the activity of this enzyme both in vitro and in vivo to provide pharmacodynamics markers of HDAC1 inhibitors in preclinical and eventually clinical development. To do this, they employed differential mass spectrometry to identify histone acetylation patterns in cells treated with a panel of known HDAC inhibitors, including those selective for HDAC1, HDAC1, and HDAC3 and HDAC1, 3, and 6, to identify the deacetylation profile selective for an HDAC1 inhibitor. They showed that the mass spectroscopy provides a unique approach to identify specific readouts of HDAC isoform selective inhibitors, which could then be used to aid in the identification and development of highly selective inhibitors of each member of the HDAC family.
Another important property and use of mass spectroscopy analysis is that it allows for identification of inhibitors of HDACs when complexed with multiple proteins, which is not easily evaluated with most other assays. This is important because in the cell, HDACs do not act as a single protein interacting with a single histone substrate but instead form multiple protein complexes. The functional outcome of those large protein complexes differs depending on the component on the complex and the histones targeted. Using mass spectroscopy and affinity capture, Bantscheff et al. 95 tested 16 different HDAC inhibitors for binding to different HDAC complexes known to form in cells and were able to discriminate the drugs by their preference for interacting with one or another complex and the downstream signaling that was subsequently affected. They profess that the approach allows them to identify novel HDAC inhibitors based on their interaction with multiple complex HDAC clusters, which serve distinct cellular functions rather than just targeting a compound to the catalytic site of an individual HDAC.
These studies are important because it suggests that mass spectroscopy has evolved to the point where it can provide moderate-throughput readouts with limiting amounts of tissue. Thus, the array of histone regulated by a specific HDAC in the context of the other HDACs in the cell can be assessed, and compounds can be screened for their ability to block that array of acetylations and not affect the actions of other HDACs.
In this context, Biocius (Woburn, MA; www.biocius.com) commercializes a RapidFire label-free mass spectroscopy technology for HDAC drug discovery, as well as for other epigenetic targets. The technology is rapid (6- to 10-s cycles) and high throughput, and it can be used to screen more than 900 000 compounds at a time. The system can identify multiple acetylation or deacetylation reactions on a single native target. Biocius has shown that it can identify multiple SIRT1 deactylations on p53 at the same time. Importantly, its drug screening technology is directly linked to chemoinformatics to allow for rapid structure-based evaluation of “hits” and of course nonhits in the same chemical series to provide a basis for chemical optimization.
Taken together, mass spectroscopy approaches may provide a more physiologically relevant method for HTS for selective HDAC inhibitors than the other approaches described above, which may translate into more clinically relevant inhibitors identified.
In recent years, there has been a revolution in the understanding of how epigenetics affects biology. However, progress in exploiting our understanding of this field into discovery and development of new therapeutics to treat disease has been relatively slow. This is somewhat perplexing because epigenetics forms the basis of all cellular diversity and, in many respects, the foundation of most if not all diseases. However, based on the development of programs put forth by the National Institutes of Health (www.nihroadmap.gov/epigenomics), this trend may be changing.
Indeed, a recent survey has indicated that there is major interest in the biopharmaceutical field to focus on the use of epigenetic-based assays for discovery of new therapeutics. 96 The prime interest is epigenetic modifiers for oncology but with additional focus on metabolic and CNS diseases. The most important focus is on developing highly selective HDAC and SIRT inhibitors because of the overwhelming evidence that these enzymes are involved in disease. Most important, there is growing availability of technologies to discover drugs targeting these enzymes, with over 12 companies already commercializing acetylation and methylation assay kits as well as development of optimized reagents essential for such assays.
The novelty of epigenetic pharmacology is that epigenetics by definition involves the targeted regulation of specific genes. It provides a basis for reprogramming the cell, from one that is abnormal to potentially one that is more normal. Most important, the field may provide the basis for designer drugs that can affect the expression of one gene out of the thousands present in the genome. Such exquisite control could have many important applications in the treatment of a large number of diseases, and because epigenetics is involved in the functioning of all organisms, epigenetic-based drugs could provide ways to target specific infectious organisms and viruses to overcome resistance, which may be one of the most important health risks the general population faces in the future.