
Abstract
Quorum sensing (QS) is a cell density–dependent signaling system that is used by bacteria to coordinate gene expression within their population. In this study, the authors describe the development and characterization of various cell-based bioassay systems for detecting QS inhibitors based on three LuxR family proteins, TraR, LasR, and the recently identified QscR. Three different gram-negative bacteria, Escherichia coli, Agrobacterium tumefaciens, and Pseudomonas aeruginosa, were employed as reporter strains to overproduce one of the aforementioned QS activator proteins and respond to inhibitors. The nine different whole-cell assay systems (three reporter strains × three QS proteins) were evaluated for their applicability and reliability by studying quantitative responses to various furanones, which are potent inhibitors of the LuxR family proteins. These results demonstrate that the cell-based bioassay systems are sensitive and reliable tools for screening of QS activators and inhibitors. This study also suggests that furanones are potentially important QS inhibitors for many LuxR-type activator proteins.
Introduction
Q
The mechanism that controls bacterial QS responses has been well studied at the molecular level. 1 A QS transcriptional activator protein forms a complex with AHLs and binds to several promoter sequences and functions as a transcription activator. Based on the interactions with AHLs, LuxR-type transcriptional activator proteins can be classified into three different classes. In Class I, the activator proteins require their cognate AHLs for the proper folding during protein synthesis and the binding between the protein and AHL is irreversible. Typically, LasR of Pseudomonas aeruginosa and TraR of Agrobacterium tumefaciens belong to Class I. 3 Class II activator proteins also require AHL for their expression in a soluble and active form, but the AHL can be removed from the AHL-protein complex reversibly. QscR, recently found in P. aeruginosa, belongs to the Class II activator protein. 4 Class III proteins, such as RhlR and EsaR, do not require AHLs for folding, and the mature proteins bind to the AHLs reversibly.5,6
Because of the importance of QS responses, a few studies on sensitive and selective bioassay methods for screening QS signaling molecules and inhibitors have been reported.7,8 The assay methods mostly rely on reporter strains, which produce luminescence or give a specific color response in the presence of signal molecules. The cell-based assay is easy and convenient to use but has been criticized for an inherent drawback, which is that it does not measure the direct interaction among an AHL, QS transcriptional activator protein, and promoter DNA sequences. In a recent study, a protein-based biochemical assay was developed with QscR as a transcriptional activator protein, and the result was compared with that from the cell-based assay. 4 Although conducted for only one transcriptional activator protein QscR, the study was interesting because both assays were carefully compared and turned out to be in a good agreement.
In this study, we report further extensive studies on cell-based assay systems that can be used for screening QS signal molecules and inhibitors. Several cell-based QS assay systems were developed and compared in the three recombinant reporter strains, P. aeruginosa PAO-216, A. tumefaciens NTL4, and Escherichia coli DH (5α). Three LuxR-type activators, two Class I proteins (TraR and LasR) and one Class II protein (QscR), were designed to regulate the expression level of β-galactosidase (LacZ) in nine reporter strains (three proteins × three strains). A variety of AHLs were tested for the ability to activate QS transcriptional activator proteins in the nine reporter strains. In addition, furanone derivatives were screened for QS inhibitors using a cell-based assay, and several furanones were demonstrated as promising template compounds.
Materials and Methods
Chemicals
4-Bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone (fimbrolide) was synthesized using a previously described procedure. 9 N-hexanoyl-DL-homoserine lactone (C6-HSL), N-octanoyl-DL-homoserine lactone (C8-HSL), N-decanoyl-DL-homoserine lactone (C10-HSL), N-dodecanoyl-DL-homoserine lactone (C12-HSL), and N-tetradecanoyl- DL-homoserine lactone (C14-HSL) were purchased from Aldrich and used as exogenous signal molecules in the bioassay. All the solvents used were either of spectral grade or distilled from glass prior to use. All other furanone compounds tested were obtained from the Korea Chemical Bank (Dae-Jeon, Korea). The structures of QS signal molecules and active furanones used in this study are shown in Figure 1 .
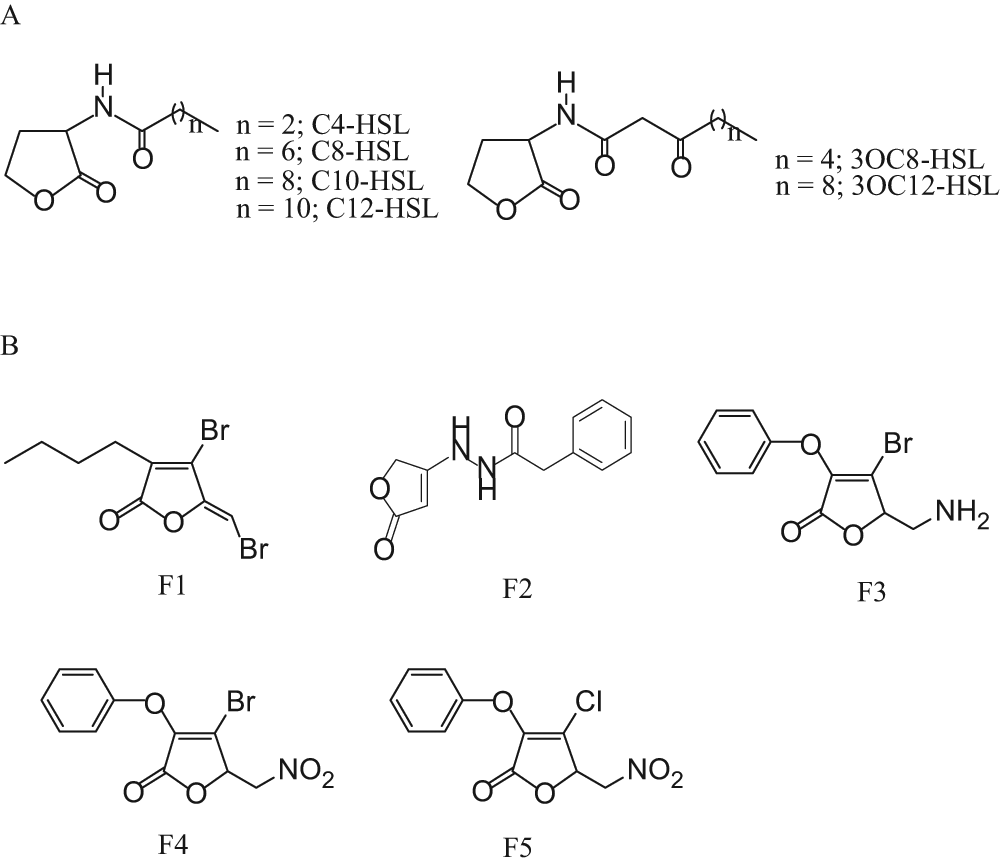
Structures of the acyl-homoserine lactones (AHLs) and furanones used in this study. (
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are summarized in Table 1 . The TraR/LasR/QscR expression vectors and the lacZ reporter vectors were used for the agonism and antagonism assays.10–13 For each reporter strain, two plasmids were incorporated. For example, TraR reporter strains harbored both pCF218 (TcR, tetracycline resistant, and KmR, kanamycin resistant) and pCF372 (SpR, spectinomycin resistant). In the TraR reporter strain, pCF218 encoded for the transcriptional activator protein TraR under the lac promoter, whereas the other plasmid pCF372 encoded for lacZ reporter under the traI promoter, which is activated in the presence of TraR-AHL complex. For LasR and QscR, the genetic elements pJN105L and pJN105Q (GmR, gentamicin resistant, araC-pBAD cloned in pBBR1MCS-5) were employed to code for LasR and QscR, respectively. Both proteins were under the control of arabinose-inducible promoter, and the addition of arabinose was required to express these proteins. The LacZ was carried on the plasmids pSC11 and pJL101 (ApR, ampicillin resistant) under the lasI promoter or PA1897 promoter, and its expression was activated by LasR-AHL or QscR-AHL complex. Recombinant strains containing the following pair of plasmids, pCF218 and pCF372, pJN105L and pSC11, or pJN105Q and pJL101, were developed by electroporation. The nine different recombinant strains (three reporter strains × three QS activator proteins) were evaluated and compared for their applicability and reliability in screening of the selected AHLs and furanones.
Strains and Plasmids Used in This Study

Cultivation of recombinant strains
Reporter strains were cultivated in a 250 mL flask (25 mL working volume) under aerobic conditions, added with AHLs (20 nM) and arabinose (0.04%) for induction of β-galactosidase activity, transferred to 2 mL Eppendorf tubes (0.2 mL), and further cultivated for expression of β-galactosidase activity. Culture temperature was fixed at 30 °C for all reporter strains. The strains expressing TraR were cultured in AT medium containing 4.5 µg/mL tetracycline and 50 µg/mL spectinomycin.11,12 The AT medium contained KH2PO4 (79 mM), NaOH (44 mM), (NH4)2SO4 (15 mM), MgSO4·7H2O (0.6 mM), CaCl2·2H2O (0.06 mM), FeSO4·7H2O (0.027 mM), MnSO4·H2O (0.0071 mM), and 0.5% glucose. Unless stated otherwise, AHLs and the inhibitors to be tested were added when the cell density reached 0.3 U of optical density at 600 nm (OD600). The strains expressing LasR or QscR were cultured in LB medium containing 10 µg/mL gentamicin and 50 µg/mL ampicillin at 30 °C. After adding AHLs, inhibitors, and/or arabinose, the cultures of 0.2 mL were distributed to 2 mL Eppendorf tubes and cultivated for 3 or 4 more hours with 250 rpm shaking. When studying the effect of induction OD on β-galactosidase activity, microbial culture taken at 0.3 OD600 was diluted with fresh medium or concentrated by centrifugation and resuspension in fresh medium. Cell concentrations (OD600) during cell growth were measured with 100 µL cultures in a microtiter plate reader (PerkinElmer, Norwalk, CT). One unit of OD600 corresponded to 0.31 mg cell mL−1 for E. coli, 0.36 mg cell mL−1 for A. tumefaciens, and 0.33 mg cell mL−1 for P. aeruginosa, respectively.
Analysis of β-galactosidase activity in reporter strains
The activity of β-galactosidase was measured according to Miller after minor modification. 14 Bacterial cells cultured with AHLs and potent inhibitors were centrifuged, washed once by Z buffer, and resuspended in the same buffer. 14 The Z buffer contained Na2HPO4·7H2O (60 mM), NaH2PO4·H2O (40 mM), KCl (10 mM), MgSO4·7H2O (1 mM), and β-mercaptoethanol (50 mM). After measuring OD600 for cell density, aliquots of 50 µL were transferred to a solvent-resistant 96-well multititer plate containing 200 µL Z buffer, 10 µL CHCl3, and 4 µL 0.1% aqueous SDS for cell lysis. This suspension was mixed via repetitive pipetting, after which the CHCl3 layer was allowed to settle. A 100 µL aliquot from the aqueous phase was transferred to a fresh 96-well multititer plate, and 20 µL of substrate, o-nitrophenyl-β-d-galactopyranoside (ONPG; 4 mg/mL in phosphate buffer), was added. The amount of cell lysates was chosen so that the degradation of ONPG is linear up to 40 min of incubation. After a 20 min incubation for color development, the reaction was terminated by the addition of 50 µL of 1 M Na2CO3. Absorbances at 420 and 550 nm were determined by a microtiter plate reader (PerkinElmer), and Miller units were calculated according to the following equation:
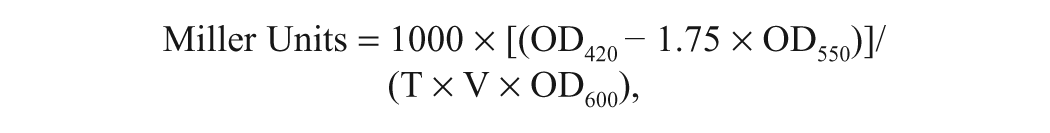
where T is the time of the reaction (min) and V is the volume of culture used in the assay (mL). A Miller unit is proportional to specific enzyme activity that is defined as µmol ONPG min−1 mg−1 protein, and the conversion factor from the former to the latter was 9.93 × 10−4 for E. coli, 9.25 × 10−4 for A. tumefaciens, and 9.49 × 10−4 for P. aeruginosa, respectively. Protein contents were determined according to Bradford assay using bovine serum albumin as protein standard. 15 The β-galactosidase activity in this article was given in Miller units. All measurements were conducted at least three times and averaged.
Results
Optimization of β-galactosidase activity measurement
The current cell-based assay method relies on quantitative and reproducible expression of lacZ gene in the presence of various AHLs and subsequent accurate determination of β-galactosidase activity in cell lysate. Therefore, the factors affecting the expression and measurement of β-galactosidase activity such as growth of reporter strains, preparation of cell lysate, color development during incubation with ONPG, and so forth need to be optimized. Because preparation of cell lysates monitored by the measurement of total protein contents and the specific rate of color development during the degradation of ONPG under given conditions (see the Materials and Methods section) were reproducible, optimization of the assay method was focused on the expression of β-galactosidase activity during incubation in Eppendorf tubes after induction.
The effect of induction OD600 on the expression of β-galactosidase activity in the reporter strains was studied, and the results with the E. coli strain harboring lasR (E. coli–lasR system) are shown in Figures 2A and 2B . A different OD600 between 0.1 and 0.7 was established in the Eppendorf tubes by diluting or concentrating cultures that were taken at 0.3 OD600, and 100 nM of 3OC12-HSL was added. As shown in Figure 2A , when 3OC12-HSL was added, the β-galactosidase activity in all cultures increased rapidly for 180 min and then rather slowly afterward. The highest activity was observed when the induction OD600 was set at 0.2 to 0.4 OD600. When induction OD was not in this range, especially above 0.5 OD600, the increasing rate of the activity with time as well as the final activity at 420 min was significantly reduced. Final cell density after 3 h incubation in Eppendorf tubes varied significantly depending on the OD at induction ( Fig. 2B ). When induction OD600 was higher than 0.5, the rate of cell growth was low. Protein synthesis in bacterial cells is related to growth rate: it is active when cells are growing rapidly and lowered when the rate is reduced. 16 The low β-galactosidase activity for the cells induced at 0.5 OD600 or above is attributed to marginal cell growth after induction. For two other reporter strains (A. tumefaciens–TraR and P. aeruginosa–QscR), similar experiments were repeated (data not shown). Different AHLs were employed depending on transcriptional activator proteins: 3OC8-HSL for TraR and 3OC12-HSL for QscR. The highest β-galactosidase activity was observed in the cells induced at 0.2~0.4 OD600, as in the case of Figure 2B , regardless of the kind of reporter strains or transcriptional activator proteins. Because the expression of β-galactosidase activity should be sensitive and reproducible in response to AHLs, 0.3 OD600 was chosen as the optimum cell density for induction in further experiments.
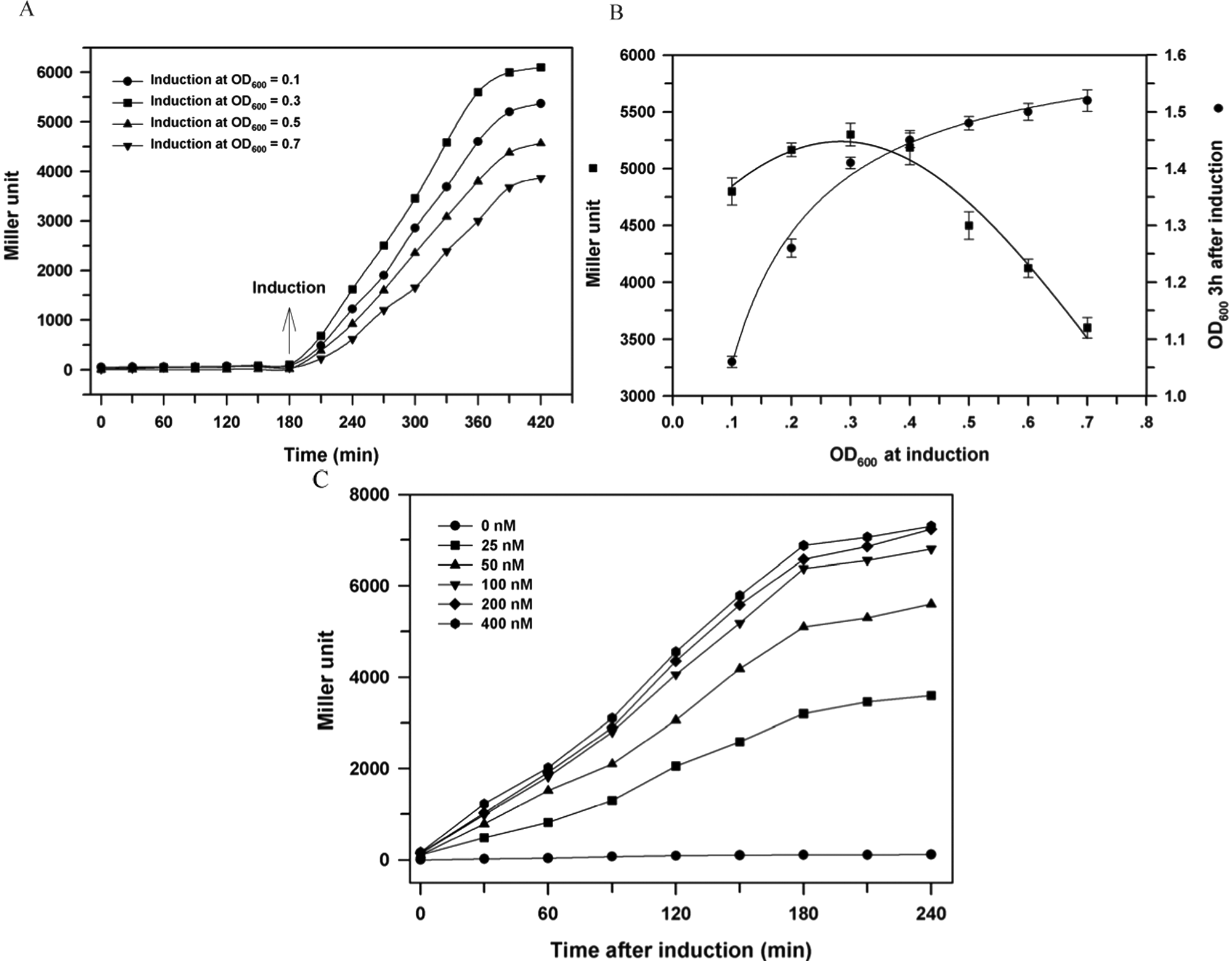
Effect of induction cell density (OD600) and acyl-homoserine lactone (AHL) concentration on β-galactosidase activity of reporter E. coli strain expressing LasR. (
Figure 2C shows the dose-response curve in the E. coli–LasR system. Induction cell density was fixed at 0.3 OD600, whereas the 3OC12-HSL concentration was varied in the range of 0 nM to 400 nM. No appreciable β-galactosidase activity was observed during cultivation of 240 min without external addition of 3OC12-HSL, confirming that the expression of lacZ is tightly regulated by 3OC12-HSL in the current reporter strain. As AHL concentration increased higher, β-galactosidase activity increased more rapidly and to a higher level. This indicates that the rate or level of lacZ expression in the current reporter strain is quantitatively controlled by the concentration of the added 3OC12-HSL. Different concentrations of 3OC12-HSL were more influential up to 100 nM in the current E. coli–LasR system, suggesting that, above 100 nM, 3OC12-HSL is almost saturated in terms of lacZ expression. Also, the increase of specific β-galactosidase activity almost stopped at approximately 180 min after induction under the current conditions. This suggests that we can get the most reliable and reproducible β-galactosidase activity when cells are harvested 3 h after induction, if induction is conducted at 0.3 OD600. A separate dose-response assay experiment was also conducted in a low 3OC12-HSL concentration range between 0 and 1.0 nM to determine the detection limit of current system, the smallest concentration at which a statistical significance (t test, p < 0.05) is observed (data not shown). The detection limit was as low as 0.1 nM for 3OC12-HSL in the E. coli–LasR system.
Similar dose-response activation assay experiments were conducted for all nine reporter strains, and the results are summarized in Figure 3 . Different AHLs were used according to QS proteins: 3OC8-HSL for TraR and 3OC12-HSL for LasR or QscR. Induction was conducted at 0.30 ± 0.05 OD600, and β-galactosidase activity was measured for the cells harvested 3 h after induction. For each transcriptional activator protein, similar dose-response curves were obtained in all three host strains. The response curve resembled Michaelis-Menten kinetics, where the β-galactosidase activity increases linearly at low AHL concentrations and gradually reaches a saturation level at high AHL concentrations. It was possible to define the half saturation constant or half maximal effective concentration (EC50) at which β-galactosidase activity becomes half of the maximum. Regardless of the kind of host strains or QS proteins, the same value of ˜25 nM was obtained. The similar dose-response curve suggests that the current reporter strains are equally reliable in detecting various AHLs. In addition, these results suggest that the effect on QS responses of AHLs transport through cell membranes and/or cytoplasm is marginal in the tested reporter strains.
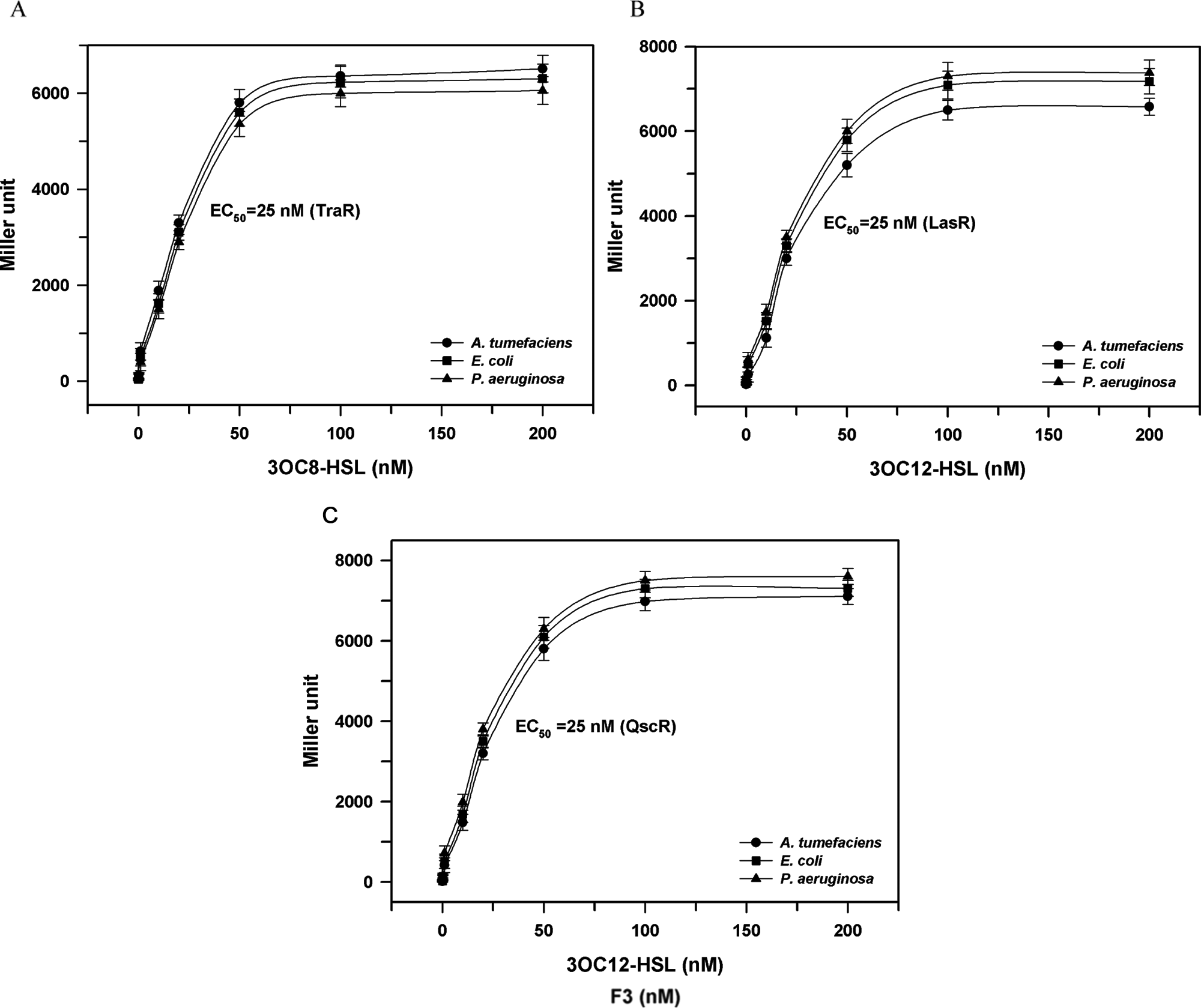
Dose-dependent activation of varying concentrations of acyl-homoserine lactones (AHLs) to quorum-sensing activator proteins and EC50 in three different hosts. The EC50 was defined as the half maximal effective concentration of AHL at which β-galactosidase activity became half of the maximum. (
Detection of various AHLs in the cell-based bioassay
The reporter strains were examined for the ability to detect various AHLs. Several AHLs, different in the fatty acyl chain length, saturation level, and/or oxidation state were tested at 100 nM against TraR/LasR/QscR in the nine recombinant strains ( Table 2 ). The response of each QS protein was most sensitive to its own intrinsic AHL among the various AHLs tested (i.e., TraR to 3OC8-HSL, and LasR and QscR to 3OC12-HSL; refer to Fig. 1 for structures). For each QS protein, all three host strains expressing the same QS strain showed almost the same sensitivity. QscR responded to a broader range of AHLs than TraR or LasR, suggesting that it is a better QS protein than LasR or TraR if a biosensor is developed to detect a broad range of AHLs. Also, it is noticed that the long-chain AHL, C14-HSL, has good activity with LasR and QscR. This suggests that C14-HSL passes through the cell membrane of the current reporter strains efficiently and is incorporated with QS activator proteins in the cell.
Activation Activity of AHLs to Quorum Sensing (QS) Activator Proteins in Different Host Strains
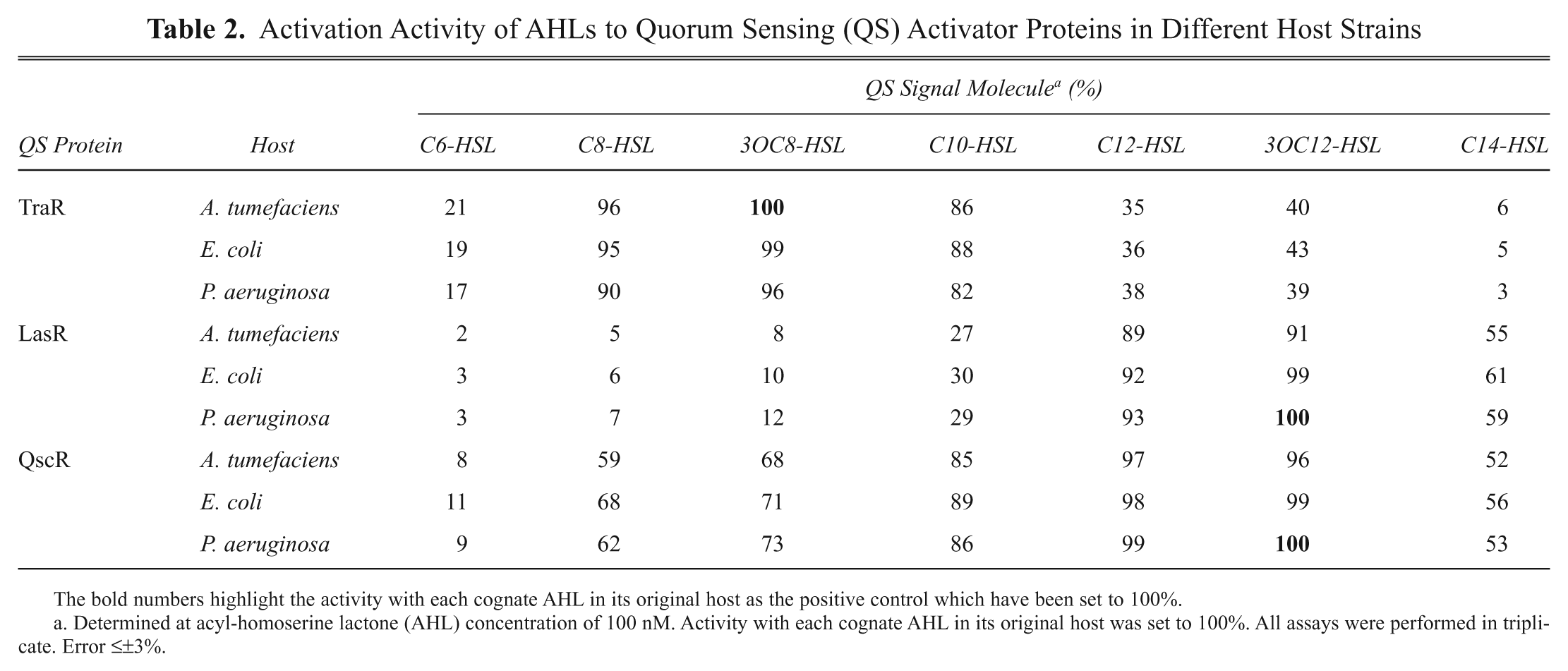
The bold numbers highlight the activity with each cognate AHL in its original host as the positive control which have been set to 100%.
Determined at acyl-homoserine lactone (AHL) concentration of 100 nM. Activity with each cognate AHL in its original host was set to 100%. All assays were performed in triplicate. Error ≤±3%.
Screening of furanones in the cell-based bioassay
The current reporter strains can be used to detect QS inhibitors. If the color response induced by AHLs is reduced in the presence of a specific chemical compound, then the chemical compound can be considered to be inhibitory to AHLs. Among various chemical compounds, furanones were chosen and tested for their inhibitory activity. A natural furanone compound, fimbrolide (F1 in Fig. 1B ), which has been isolated from a marine red macro alga Delisea pulchra, has been reported to show high QS inhibitor activity in a swarm mobility assay with Proteus mirabilis. 17 In our previous study, fimbrolide also showed strong inhibition on TraR-mediated QS gene expression in A. tumefaciens.11,12
For the first-round screening, 120 furanone compounds obtained from the Korea Chemical Bank (Dae-Jeon, Korea) were tested at 400 nM (
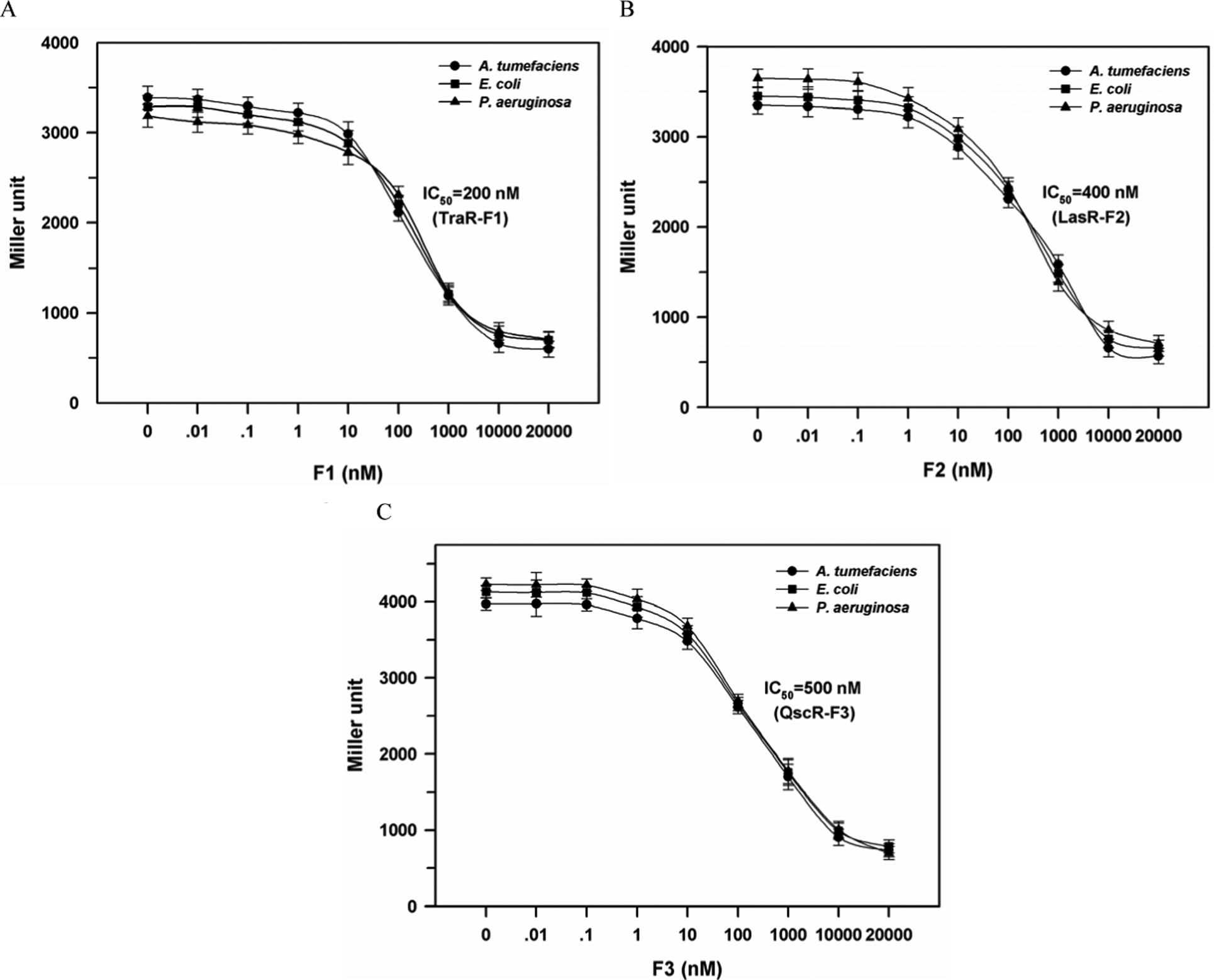
Dose-dependent inhibition curve and IC50 for active furanones. The IC50 was defined as the concentration of inhibitor at which β-galactosidase activity became half of the positive control determined with 20 nM AHL but without addition of the inhibitor. See
Fig. 1
for the structure of F1, F2, and F3. (
Inhibition of five furanones on the QS response was tested for the nine reporter strains ( Table 3 ). The AHLs were fixed at 20 nM, whereas furanones were at 2 × 104 nM, which is a 1000-fold molar excess of AHLs. The extent of inhibition by each furanone was significantly different depending on the kind of QS protein, and the most active inhibitors were identified as F1 for TraR, F2 for LasR, and F3 for QscR, respectively. However, different strains having the same QS protein did not show much difference in the extent of inhibition. For the three most active furanone compounds, dose-dependent experiments were performed with nine recombinant reporter strains ( Fig. 4 ). The concentration of AHLs (3OC8-HSL for TraR and 3OC12-HSL for LasR and QscR) was fixed at 20 nM, whereas furanone concentrations were varied in the range of 0.01 to 2 × 104 nM. The 50% inhibition concentrations (IC50) were determined as 200 nM or 10-fold molar excess for TraR, 400 nM or 20-fold molar excess for LasR, and 500 nM or 25-fold molar excess for QscR. These results indicate that the inhibition is mostly affected by interaction between QS proteins and AHLs or furanones but not by host strain.
Inhibition of Furanones against AHL Activation for QS Proteins in Different Host Strains
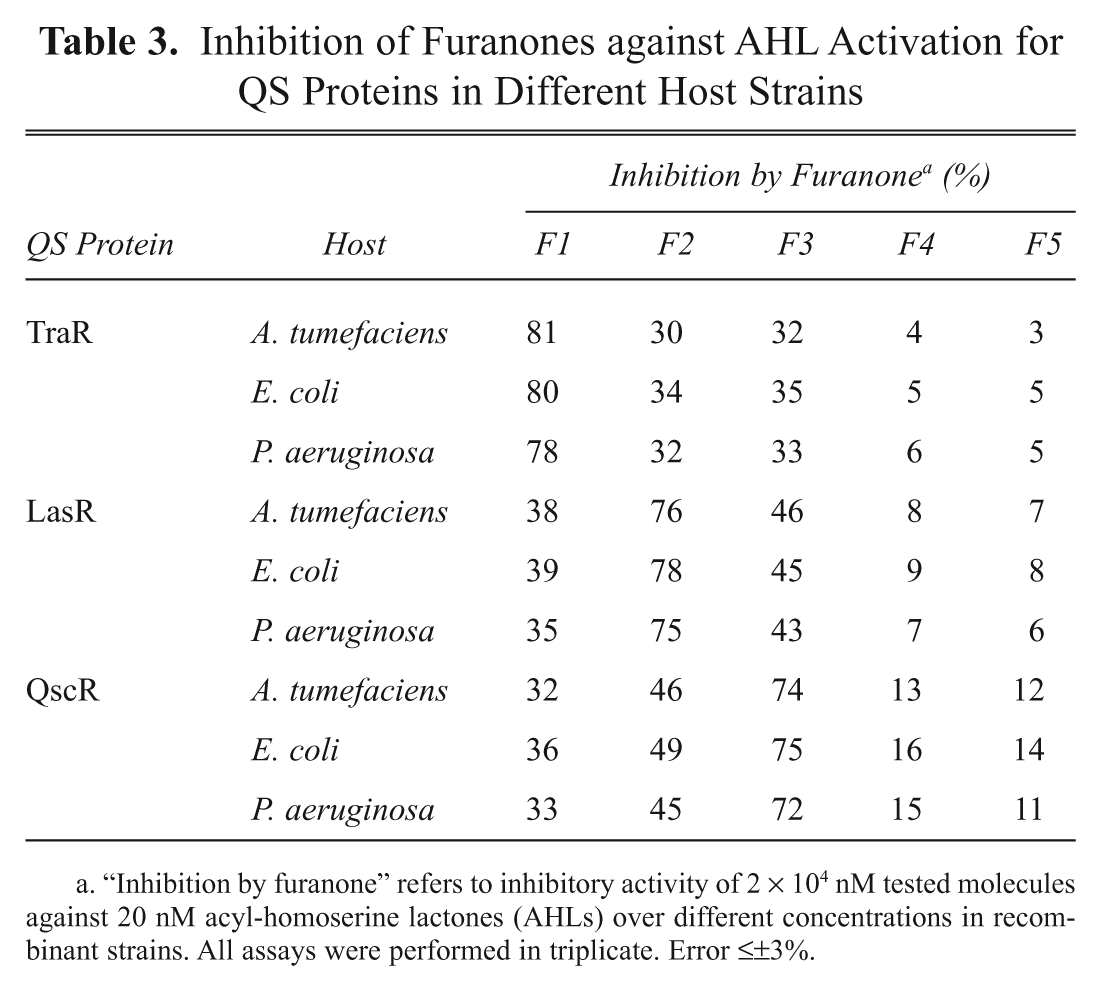
“Inhibition by furanone” refers to inhibitory activity of 2 × 104 nM tested molecules against 20 nM acyl-homoserine lactones (AHLs) over different concentrations in recombinant strains. All assays were performed in triplicate. Error ≤±3%.
Discussion
The cell-based assay system for detecting bacterial QS signaling molecules or inhibitors has been criticized because it does not measure the direct interaction between the target compound and the QS activator proteins. Efforts to develop a protein-based biochemical assay were not successful for most QS activator proteins because the proteins in the AHL-free form could not be obtained. It is believed that QS activator proteins, especially Class I LuxR-type proteins, incorporate their corresponding AHLs during the translation process and bury them deep inside. 3 Therefore, the AHL molecules cannot be removed after the purification without destroying the protein structure. 18 Also, the expression of QS proteins without a proper AHL resulted in the formation of inclusion body, which could not be refolded into the active form. Considering that the protein-based biochemical assay cannot be developed for many QS activator proteins, the consistency and reliability of the cell-based assay is important. Further verification with other QS activator proteins and host strains need to be performed to increase the reliability of the cell-based assay.
A large number of compounds have been reported as QS inhibitors, but the universal QS inhibitors that are active for many diverse QS proteins have not been developed yet. The inhibitors for QS activator proteins reported so far are very diverse in their molecular structure and mass. This is partly due to the difference in the structure of binding pockets in different QS activator proteins. According to protein structure analysis, both LasR and TraR have different pocket sizes, which enable them to accommodate their signal molecules precisely, shown by a bigger binding pocket in LasR (~670 Å 3 ) and smaller binding pocket in TraR (~440 Å 3 ). The current study showed that furanone compounds have inhibitory activity for many LuxR-type QS activator proteins. Although the level of the activity was different depending on the kind of furanones and proteins, furanone compounds collectively have the potential to be developed as a template inhibitor for many LuxR-type activator proteins.
We have developed and evaluated nine recombinant strains to screen for QS activators and inhibitors. These cell-based QS screening systems have been proven sensitive and reliable when applied to screening of QS inhibitors from a library of synthetic furanone compounds. Some furanones, such as F1, F2, and F3, strongly inhibited QS responses in reporter strains. Furanone compounds can be considered a promising template for developing an additional chemical library for QS inhibitors of LuxR-type QS activator proteins.
Footnotes
Acknowledgements
This work was financially supported by the Ministry of Education, Science and Technology through Advanced Biomass Research Center (Global Frontier Program), KAIST and Brain Korea 21 program, Pusan National University, Korea.
The authors declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
The authors received no financial support for the research and/or authorship of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.