
Abstract
This study investigated the use of large-scale transiently transfected cryopreserved cells for medium-throughput cellular screening. The data generated indicated that preprepared transiently transfected cryobanks can be used for cell-based assays and in fact can greatly enhance the consistency of data generated by cellular screens. In addition to this, a generic enzyme-linked immunosorbent assay method was designed that introduced a c-Myc tag to four different targets and allowed all four cell assays to be run using a standardized process. These process improvements yielded cost savings and greatly reduced the required resource, as well as reducing timelines for developing cellular assays.
Introduction
T
To this aim, we introduced an enzyme-linked immunosorbent assay (ELISA) method that identifies inhibitors of a c-Myc-tagged target kinase by measuring a decrease in the phosphorylation of a transiently overexpressed target following pretreatment with compound. This generic capture ELISA procedure is based on the recognition of the c-Myc tag rather than the antigen itself, meaning that antigen-specific antibodies are not required for the ELISA. 1 The use of the anti-c-Myc capture antibody can allow detection of several antigens tagged with a c-Myc polypeptide on the c-terminus of the antigen required for testing. Previous studies have shown that using a capture ELISA process can offer comparable if not higher sensitivity than direct ELISA methods. 2 In this investigation, four different antigens were assessed using the c-Myc tag. In addition to this, the ELISA method used to detect the decrease in phosphorylation of these four targets was standardized, to allow a reduction in development timelines of any new target investigated.
The four targets chosen for this standardization process were all kinases. Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase ubiquitously expressed in cells and localized to focal adhesions. FAK plays a key role in the turnover of adhesion complexes and has both kinase-dependent and kinase-independent functions. 3 Pancreatic endoplasmic reticulum eIf2α kinase (PERK) is a transmembrane kinase that phosphorylates the translation initiation factor eIf2α and has an important role in monitoring the quality of protein folding in the endoplasmic reticulum (ER). The phosphorylation of eIf2α results in the reduction of cellular protein synthesis and thus affects the number of proteins entering the ER. 4 Dual-specificity tyrosine–(Y)-phosphorylation regulated kinase 1B (DYRK) is a member of a conserved family of serine/tyrosine kinases that are activated by intramolecular tyrosine phosphorylation. DYRK 1B is involved in mediating differentiation in skeletal muscle and neuronal cells. 5 The fourth target assessed in this study was bone marrow X-linked (BMX) tyrosine kinase, which plays an important role in mediating integrin signaling in endothelial and epithelial cells. 6
Part of this work to standardize the assay process involved the use of cryopreserved cells, which has been shown in previous studies to generate data that are reliable and reproducible for a variety of cell lines. 7 For most cellular screening assays, continuous cell culture is often required, with weekly transfections of these cell lines carried out where overexpression is necessary. This process can introduce variability into cellular screening because there is the potential of passage-related variation, and any change in handling procedures and conditions (incubation timings and temperatures, etc.) could easily affect cell behavior. It has been shown in previous studies that large-scale production of cryopreserved cells can offer several benefits over routine culture. 8 These include reproducibility and reliability, as each vial will provide the same number of cells at the same passage, thus reducing the potential impact of passage variation. Cryopreserved cells also increase the overall flexibility of cellular screening as they are convenient and easy to use because they are available whenever they are required. They also provide cost and time savings as there is no need to perform weekly cell culture or transfections. For this study, we investigated the benefits of using large-scale transiently transfected cryopreserved cells.
In certain cell lines, the process of cryopreservation has been shown to induce some cellular damage to certain cell lines, which can result in the excessive shrinkage and even death of cells. 9 The processes used for cryopreservation have been improved to reduce cell damage and enhance recovery. In addition to this, the use of transient transfections over stable cell lines has been investigated as transfection reagents can yield transfection efficiencies high enough to perform functional assays. For cell assays using small-scale transfections, proprietary transfection reagents are often used. However, for large-scale/high-density transfections required for screening, these reagents are often costly, can create handling issues, and can sometimes prove cytotoxic when concentration is scaled equivalently. Work has been carried out to reduce the cost and handling issues associated with large-scale transfections and to make the process more efficient. Polyethylenimine (PEI) provides a cost-effective transfection reagent that we have found to be less cytotoxic in our assays.
By combining the use of readily available cryobanks with standardized processes, cellular screening can become more efficient. This study shows that optimizing the full process of a cellular assay from cell plating through to the end measurement can give more reliable and consistent data, and the procedures used are generic and user-friendly.
Materials And Methods
The 384-well tissue culture plates and 10-layer CellSTACKS were supplied by Corning Incorporated (Corning, NY), and the 384-well high-bind ELISA plates were supplied by Greiner Bio-One (Monroe, NC). The 384-well poly-D-lysine-coated tissue culture plates for HEK 293 cell plating were purchased from Becton Dickinson (BD) Biosciences (Franklin Lakes, NJ). Labcyte, Inc. (Sunnyvale, CA) supplied the 384-well source plates for acoustic liquid dispensing.
Dulbecco’s modified Eagle’s medium (DMEM) and F12 medium were supplied by Gibco Invitrogen Corp. (Life Sciences Limited, Paisley, Scotland). Fetal calf serum (FCS) and bovine serum albumin (BSA) were purchased from Sigma-Aldrich Company Limited (Dorset, England). Opti-MEM and Lipofectamine 2000 reagents used for transfections were supplied by Invitrogen, and PEI, linear 25 kDa (cat. #23966), was purchased from Polysciences (Warrington, PA).
Lysis buffer comprised 25 mM Tris/HCl, 3 mM EDTA, 3 M EGTA, 50 mM NaF, 2 mM sodium orthovanadate, 0.27 M sucrose, 10 mM B-glycerophosphate, 5 mM pyrophosphate, and 0.5% (v/v) Triton X-100. Complete protease inhibitor tablets (Roche, Basel, Switzerland) were dissolved in lysis buffer 20 min before use. Plate washer solution was prepared by adding 0.05% polysorbate and 10 ppm of ProClin 300 (Sigma-Aldrich) to phosphate-buffered saline (PBS)/A solution.
FAK primary antibody was purchased from BioSource (Camarillo, CA), PERK phospho–primary antibody was supplied by Santa Cruz Biotechnology (Santa Cruz, CA), and DYRK phospho-antibody was manufactured in-house. Anti-rabbit IgG–horseradish peroxidase (HRP) linked antibody and the Myc tag (9B11) capture antibody were purchased from Cell Signaling Technology (Danvers, MA). BMX used streptavidin-HRP supplied by GE Healthcare (Piscataway, NJ) and anti-phosphotyrosine clone 4G-10-Biotin conjugate from Millipore (Billerica, MA). QuantaBlu fluorogenic peroxidase substrate kit was supplied by Thermo Scientific (Rockford, IL).
The FAK, PERK, and DYRK 1B genes were codon-optimized and synthesized by GeneArt (Invitrogen, Paisley, UK) to include a C-terminal c-Myc tag. These sequences were then individually subcloned into pcDNA3.1 from Invitrogen. The resultant constructs contained the appropriate coding region with a downstream c-Myc tag sequence under the control of a cytomegalovirus (CMV) promoter. The BMX gene was subcloned from an Invitrogen Genestorm clone (H-X83107M) into pGEN-IRES-neo (proprietary AstraZeneca vector). Again, the resultant construct contained the BMX coding region with a downstream c-Myc tag sequence under control of a CMV promoter.
Cell culture
FAK and PERK used the epithelial-like human embryonic kidney cell line (HEK 293) cultured in DMEM supplemented with 10% FCS; for FAK, the media were also supplemented with 25 mM HEPES. DYRK used the fibroblast-like African green monkey kidney cell line Cos-1 with the growth medium phenol red–free DMEM supplemented with 10% FCS, and BMX used CHO K1 cells (an epithelial-like Chinese hamster ovarian cell line) grown in F12 media supplemented with 10% FCS.
Fresh transient transfections
The appropriate cell lines were cultured biweekly, and all were used in assays when the cells reached ~80% confluency. All cell lines were transiently transfected using DNA (each target was tagged with a c-Myc construct) diluted to 20 µg/mL using Opti-MEM medium mixed with Lipofectamine 2000 reagent prepared in Opti-MEM (FAK used a dilution factor of 1/53 Lipofectamine in Opti-MEM, PERK used 1/34, DYRK used 1/55 dilution, and BMX required a dilution of 1/35). For all assays, the DNA preparation and Lipofectamine solution were mixed (v/v) and allowed to incubate at room temperature for 20 min before being mixed gently with the cells at a ratio of 5:1 (cells:mix) for all cell lines (FAK at a cell density of 1.5 × 105 cells/mL, PERK and BMX at a density of 3 × 105 cells/mL, and DYRK at a density of 1.2 × 105 cells/mL).
Lipofectamine 2000 transfections
All cell lines were grown in 10-layer CellSTACKS (Corning) using the appropriate growth media until they were 80% to 90% confluent. Lipofectamine 2000 and DNA were diluted in Opti-MEM at the same concentration and ratio as used in fresh transfections. The two dilutions were combined and allowed to incubate at room temperature for 20 min to allow transfection complexes to form. The media from the CellSTACK were removed, and fresh media containing the transfection mix were added to the cells and allowed to incubate for 4 h to allow transfection to occur. The cells were then harvested and diluted to 1 × 107 viable cells/mL using growth media containing 20% (v/v) FCS. Once a homogeneous cell suspension was achieved, an equal volume of freeze medium (90% FCS, 10% DMSO) was added. The suspension was quickly dispensed into cryovials and frozen at −80 °C for 1 h before removing to vapor phase N2 vessels.
PEI transfections
Cell culture procedures were used as described in the previous section. The transfection mix contained PEI diluted in Opti-MEM and DNA construct diluted in Opti-MEM. For HEK 293 cells, 1.6 mg DNA and 2 mg PEI were used to transfect 1 × 108 cells. For COS-1 cells, 2 mg DNA and 1 mg PEI were used to transfect 1 × 108 cells. The DNA dilution was added to the PEI dilution with continuous mixing for 2 to 5 min to allow complexes to form. The transfection complexes were added to the cells (at a density of 2.5 × 106 cells/mL) and incubated for 3.5 h at 37 °C, 5% CO2, 140 rpm in an orbital incubator. The cell suspension was removed, and the cells harvested by centrifugation. At this point, the cells were prepared for cryopreservation exactly as described above for the Lipofectamine 2000 transfection.
Cell seeding
For assaying, the required number of cryovials was removed from storage and thawed at 37 °C for ~1 min. Cells were diluted with 10 mL of the appropriate media and harvested by centrifugation at 1000 rpm for 5 min to remove DMSO in freeze mix.
For assaying, the fresh transfections and cryovials were diluted to have the same final density of cells/well. For FAK and PERK, the cell suspensions were plated out at 40 µL/well using a WellMate (Matrix; Thermo Scientific) into 384-well poly-D-lysine-coated plates at final cell densities of 1.25 × 105 cells/mL and 2.5 × 105 cells/mL, respectively. DYRK and BMX cell suspensions were plated out at 40 µL/well using a WellMate into 384-well tissue culture plates at final cell densities of 1.17 × 105 cells/mL and 2.5 × 105 cells/mL, respectively.
Standard tissue culture conditions were used for all cell lines, grown at 37 °C in 90% relative humidity and 5% CO2 in a rotating incubator (LiCONiC Instruments, Boston, MA) 12 to 20 h prior to performing the assays.
ELISA plate preparation and compound treatment
The anti-c-Myc 9B11 capture antibody was coated onto 384-well high-bind plates (Greiner Bio-One) at 1 out of 800 dilution (diluted in PBS) using a WellMate and incubated for 12 to 24 hours at 4 °C. The ELISA plates were subsequently blocked using 3% (w/v) BSA in PBS/A for 2 to 3 h at room temperature.
DMSO (maximum signal) and compounds providing minimum signals for each assay were prepared at the appropriate concentration in DMSO in acoustic-certified microplates (Labcyte, Inc.). Compounds for IC50 analysis were solubilized at 10 mM in DMSO and then serially diluted using a Hydra liquid handler (Matrix; Thermo Scientific) to give four 1:100 dilutions in the microplates. Cell plates were dosed using a Labcyte Echo 555 acoustic dispenser. 10 The cells were dosed over a 12-point range from 30 µM down to 0.00003 µM to calculate the compounds’ IC50s. The plates were then incubated for 2 to 3 h (except DYRK, which required a 5-h incubation period after dosing) at 37 °C, 90% relative humidity, and 5% CO2 in a rotating incubator. The cells were then lysed by adding 40 µL/well of lysis buffer using a WellMate dispenser. After a 10-min incubation on ice, 15 µL of lysate was transferred onto the preblocked ELISA plates using a PlateMate plus liquid handler (Matrix; Thermo Scientific). The additional cell lysates could also be transferred onto preblocked ELISA plates and used to measure total levels of required target (data not shown). The ELISA plates were then incubated for 2 to 4 h at room temperature or 18 to 24 h at 4 °C, to allow binding of the c-Myc-tagged antigen.
Generic ELISA procedure
A generic ELISA method was created to run several assays. The process is as follows:
Wash: the plates were washed three times with 400 µL of plate wash solution using a 384-well Platewasher (Tecan, Männedorf, Switzerland).
Reagent addition: 20 µL/well primary antibody was added (either a biotinylated-4G-10 [1/2000 dilution] or a target-specific phospho or total antibody [dependent on assay]) using a WellMate.
Incubation: the plates were incubated at room temperature for 60 min.
Wash: the plates were washed three times with 400 µL of plate wash solution.
Reagent addition: 20 µL/well secondary antibody was added (anti-rabbit IgG-HRP linked [1/4000 dilution] or streptavidin-HRP linked [1/4000 dilution]) using a WellMate.
Incubation: the plates were incubated at room temperature for 60 min.
Wash: the plates were washed three times with 400 µL of plate wash solution.
Reagent addition: 20 µl/well QuantaBlu substrate solution was added.
Incubation: the plates were incubated at room temperature for 60 min.
Reagent addition: 20 µL/well QuantaBlu Stop solution was added.
Analytical methods and data analysis
The plates were analyzed using a Safire II platereader (Tecan) with a fixed fluorescence setting at excitation wavelength 340 nm (10-nm bandwidth) and emission wavelength 465 nm (10-nm bandwidth). The method used an optimal gain calculated from the first plate in the stack.
There were several measures of data quality assessed for this study. The window between the mean maximum (max) and mean minimum (min) fluorescent signals was used to calculate a Z′ factor 11 for every plate tested. The coefficient of variation (CV) for the controls was also assessed for each plate of compounds tested. In addition, several reference compounds were run in each assay to show a correlation with the historical data. The IC50s for each compound tested were calculated by creating a response curve using the results from the 12-point dose range (30–0.00003 µM).
Results
Each of the individual assays was already developed with a c-Myc tag prior to this study, and the validation data set for each assay as a modular method is not included in this report. The assays were adjusted to create a more generic process by altering incubation times of lysate addition, as well as reagents used and the possibility of adjusting assays to use the same semiautomated processes (i.e., using the same wash procedure and buffers). There was no significant impact on the pIC50s of the standard compounds generated for the assays by making these adjustments, and the correlation between pIC50s of 2- versus 24-h incubation was very good (data not shown).
To help create an optimized process, each of the assays was tested using a bank of prepared cryopreserved cells in comparison with freshly prepared transfected cells. Figure 1 shows the correlation of pIC50s between all four targets for the investigation of each cryobank. FAK and PERK show the best correlation between the cryopreserved cells and fresh cells (R2 = 0.94 and 0.95, respectively), whereas DYRK and BMX have a lower correlation (R2 = 0.75 and 0.68, respectively). This is still acceptable, however, because there was no significant shift in compound potency seen in the assays. Table 1 also shows that the use of cryovials can maintain the low CV values (excluding the minimum results for PERK and DYRK—fresh, 14.0% and 11.9%, to cryo, 32%) and has no significant impact on Z′ values obtained. The results for FAK and DYRK showed an increase in Z′ values when using a cryobank (0.8–0.85 and 0.29–0.41, respectively), whereas results for PERK and BMX did show a slight decrease in Z′ values (0.63–0.53 and 0.57–0.52), but this could be considered variability seen from assay to assay. All Z′ were deemed acceptable for use in cellular screening (cutoff 0.3), and all targets were further investigated using cryopreserved cells.
Comparison of Freshly Prepared Transient Transfected Cells and the Preprepared Transiently Transfected Cryopreserved Cells
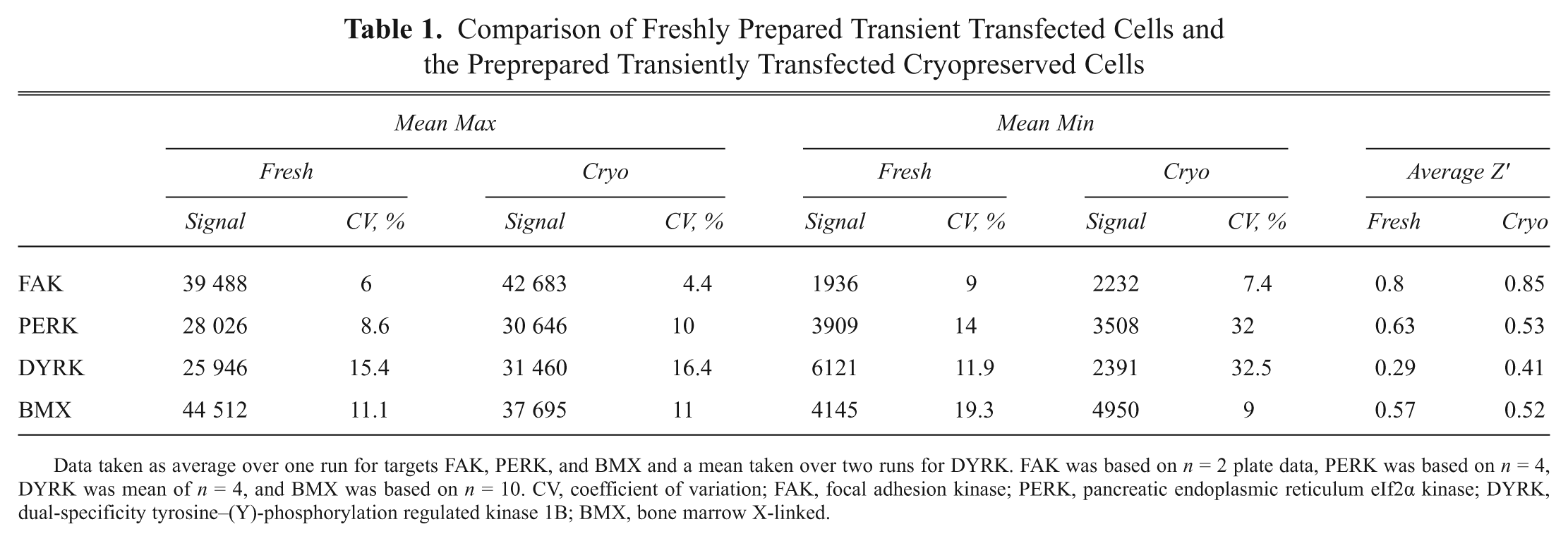
Data taken as average over one run for targets FAK, PERK, and BMX and a mean taken over two runs for DYRK. FAK was based on n = 2 plate data, PERK was based on n = 4, DYRK was mean of n = 4, and BMX was based on n = 10. CV, coefficient of variation; FAK, focal adhesion kinase; PERK, pancreatic endoplasmic reticulum eIf2α kinase; DYRK, dual-specificity tyrosine–(Y)-phosphorylation regulated kinase 1B; BMX, bone marrow X-linked.
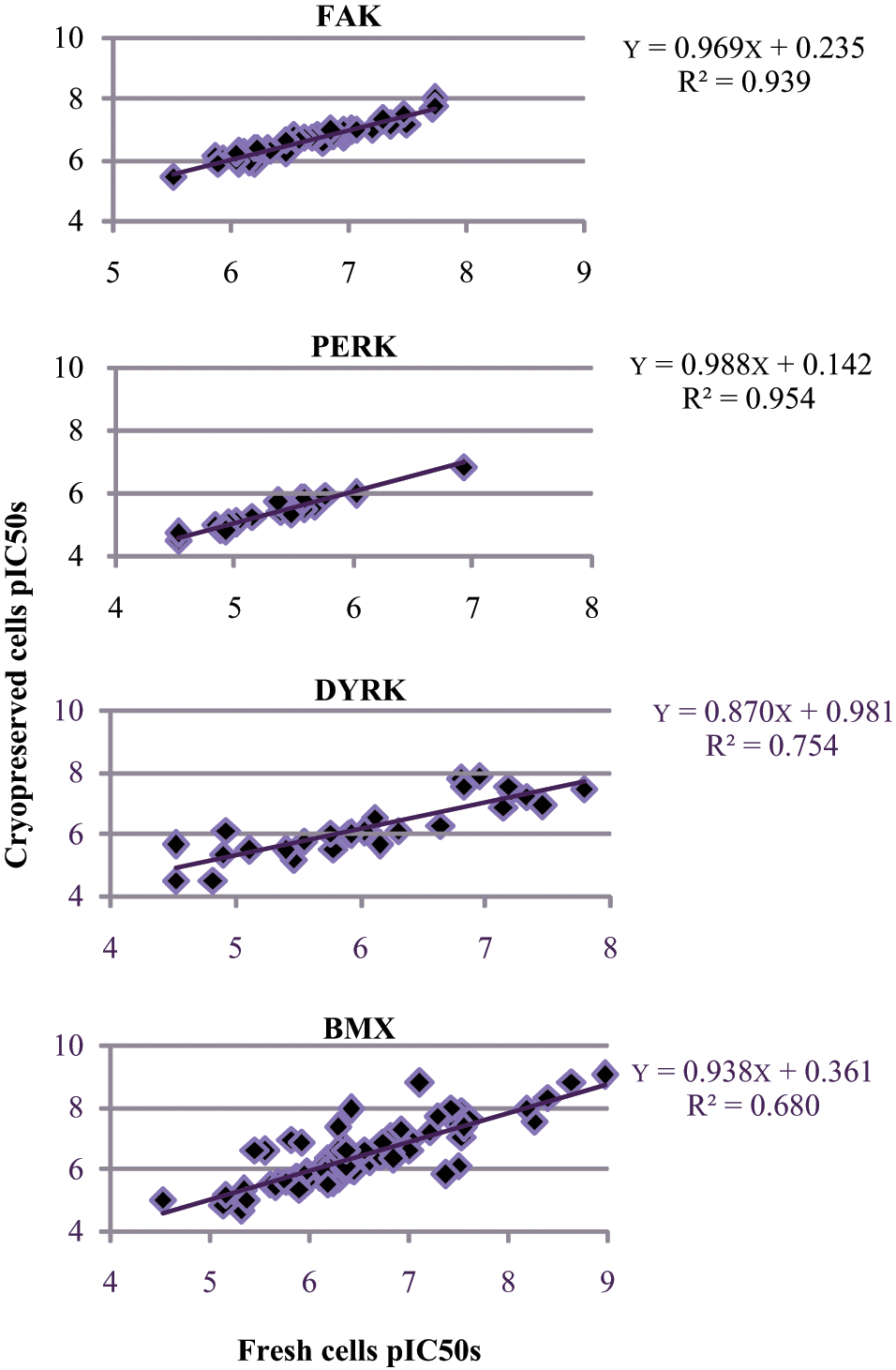
Correlation of pIC50s for selective compounds for fresh transfected cells versus preprepared cryobank for all four targets. Data set was collected from validation runs of n = 3 with minimum of 40 pIC50s generated. FAK, focal adhesion kinase; PERK, pancreatic endoplasmic reticulum eIf2α kinase; DYRK, dual-specificity tyrosine–(Y)-phosphorylation regulated kinase 1B; BMX, bone marrow X-linked.
Additional work was also carried out to compare the two transfection reagents Lipofectamine 2000 and PEI for FAK. The results ( Fig. 2 ) indicated there was no significant impact of moving away from Lipofectamine 2000 as the pIC50 correlation indicated no shift in potency (R2 = 0.89).
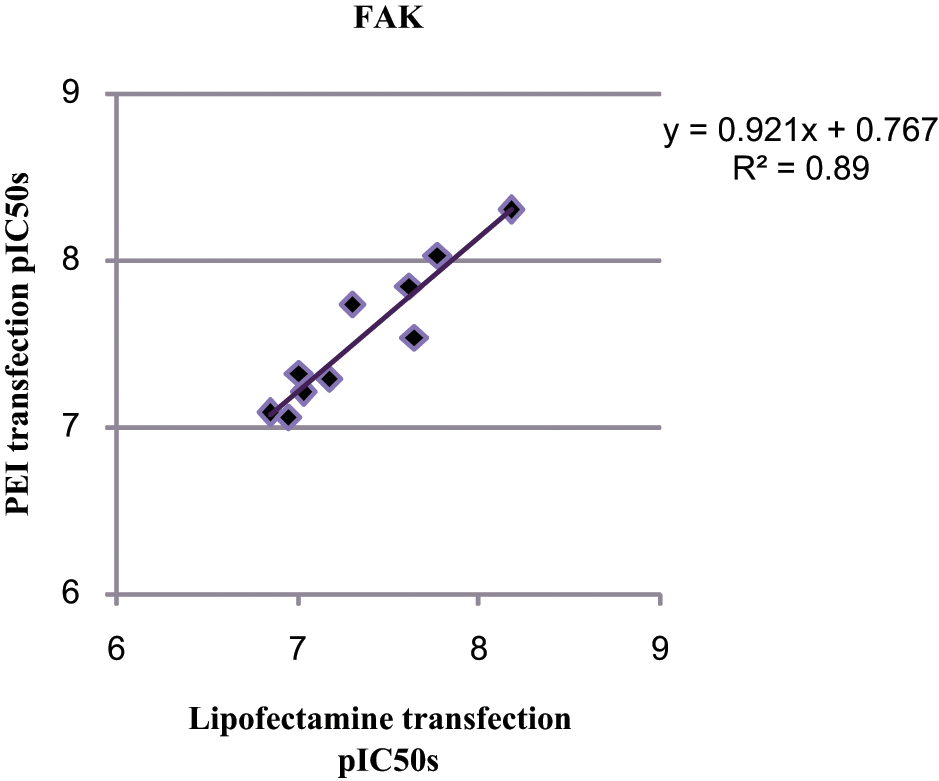
Comparison of different transient transfection reagents (Lipofectamine 2000 and polyethylenimine [PEI]) on the generation of pIC50s for focal adhesion kinase (FAK) only. An n = 3 validation set was carried out and 12 pIC50s of various compounds were generated in each run.
Assay performance
The intra-assay results obtained with cryopreserved cells for FAK, DYRK, and BMX are shown in Figure 3 . The data show that FAK is very consistent over one run with the Z′ remaining around 0.84 to 0.85, and the window between the maximum and minimum fluorescent signals also remains constant with a fold difference of 19 (approx. 42 680–2200). The targets DYRK and BMX show a higher variability when using the fresh cells over one run when compared with the cryopreserved cells. The Z′ for DYRK remains consistent over a run for the cryopreserved cells but does vary slightly for freshly prepared cells (cryopreserved, 0.4–0.41; fresh, 0.3–0.47). The minimum signal for the fresh cells is also higher than the cryopreserved cells (9000 vs 2300). BMX shows a similar story to DYRK, where the fresh cells appear to show more variability (0.13–0.42) when compared with the cryobank (0.43–0.68). The results for BMX show a marked improvement using the cryopreserved cells as the assay has a much higher background using freshly transfected cells (17 000 vs 5000 for cryopreserved cells).
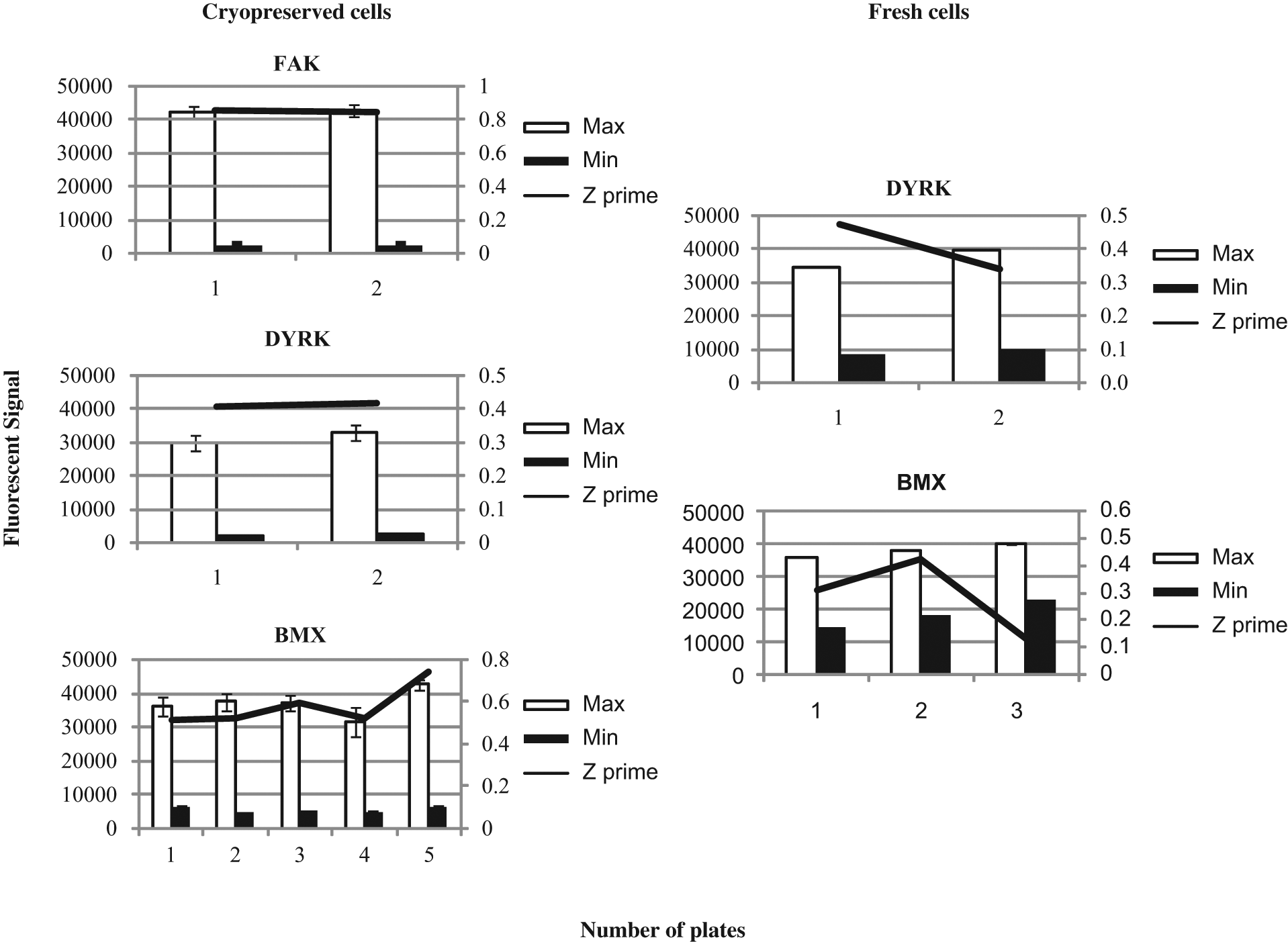
Intra-assay performance of freshly prepared transfections versus transiently transfected cryopreserved cells. Standard deviations are based on a minimum of n = 17 maximum and minimum controls on each plate run for every target investigated, and a minimum of two plates per assay were run for this evaluation. FAK, focal adhesion kinase; PERK, pancreatic endoplasmic reticulum eIf2α kinase; DYRK, dual-specificity tyrosine–(Y)-phosphorylation regulated kinase 1B; BMX, bone marrow X-linked.
The interassay data generated for DYRK and BMX are shown in Figure 4 . The results show that from run to run, the cryopreserved cells are more consistent than the fresh cells for both targets. The results for DYRK show the most significant improvement from using cryovials over the fresh transfections. The Z′ values for fresh cells varied from 0.13 to 0.47 from week to week, and this is primarily due to the changes in fluorescent signal for both maximum and minimum treatments (maximum range, 21 000–40 000; minimum range, 4371–9819). In comparison, the cryobank gave a tighter range of values for both maximum and minimum signals (max range, 23 000–38 000; minimum range, 2300–6400), which resulted in less variable Z′ being produced (0.3–0.45). BMX showed a similar comparison of fresh and cryovials to DYRK; however, there were less runs carried out for BMX using fresh cells. The fresh transfections gave variable Z′ values (0.29–0.57), and although the maximum signal was consistent between runs, the minimum signal background for one of the runs was very high (18 000 vs 4200) and resulted in a low Z′ value. The CHO K1 cryobank used for BMX gave reproducible results (maximum range, 321 000–43 000; minimum range, 5000–6900) and improved Z′ over the fresh cell (0.45–0.74).
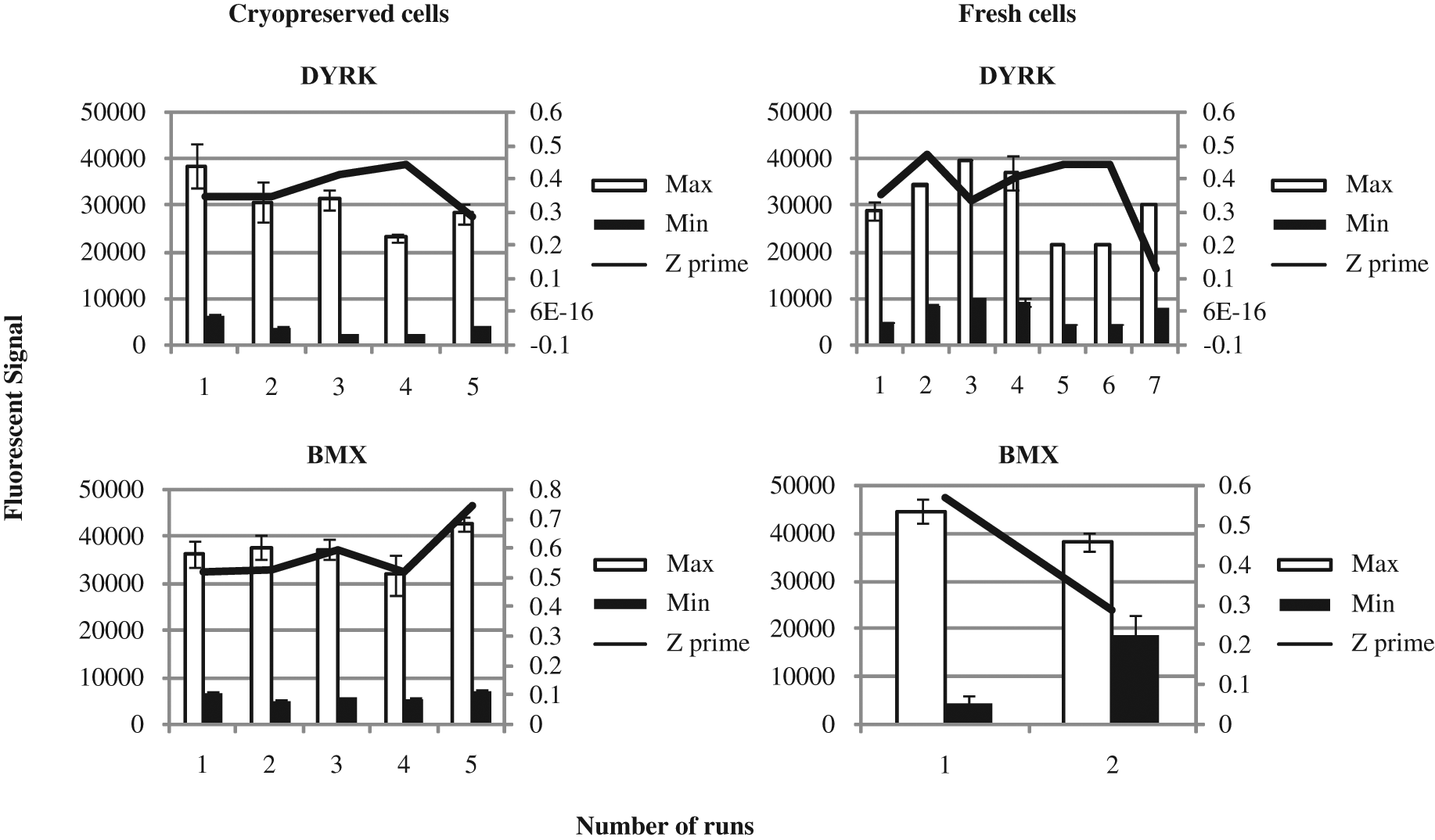
Interassay performance for freshly prepared transfections versus transiently transfected cryopreserved cells. Standard deviations are based on a minimum of n = 17 maximum and minimum controls on each plate run for every target investigated, and a minimum of two plates were investigated in each run. DYRK was investigated over five validation runs for cryopreserved cells and over seven runs for fresh cells. BMX was an evaluation of fresh cells over two validation sets and was run using five runs for cryopreserved cells. DYRK, dual-specificity tyrosine–(Y)-phosphorylation regulated kinase 1B; BMX, bone marrow X-linked.
Discussion
An ideal cellular assay is simple, robust, and reproducible and should be easy to automate, thus allowing for a higher throughput. By introducing a generic method that can be used for various targets and successfully standardizing a semi-automated process, cellular screening can become more efficient, as shown in this study.
Converting the assay process to use a cryobank instead of fresh cells means that the data could be made more consistent run to run because every cryovial tested was transfected with the same DNA mix in each vial. Culturing fresh cells for use in the assay can create variability because each cell line can behave differently depending on passage number, therefore resulting in differences in the growth and health of cells. The results of this investigation showed that using cryopreserved cells instead of freshly transfected cells had no significant impact on the data generated for PERK and gave an improvement in performance for FAK, DYRK, and BMX. The data generated during the study indicated that using a cryobank of cells can reduce variability, enhance overall performance, and provide a more efficient way of screening cellular assays.
Several cell lines were assessed for their feasibility to be used as a cryobank, as the ability of the particular cell line to recover after a freeze/thaw process could result in differences in behavior of each assay and therefore the data generated. For example, BT474 and HT29 cells are not feasible for use in this process as they took too long to recover from frozen (data not shown). It was found in this investigation that for DYRK, the COS-1 cells took longer to revive from frozen and had to be incubated postrecovery for no less than 18 h before performing an assay. In contrast, the cell line for BMX (CHO K1) recovered very quickly from the freeze/thaw process, and the optimum seeding time was 12 h before use in assay (data not shown). The HEK 293 cells for FAK and PERK revived quickly but had to be seeded in poly-D-lysine-coated plates to help them adhere 12 to 16 h before use in the assay. These differences in cell behavior mean that although the majority of the process can be designed to be generic, the cell seeding has to be optimized for each individual cell line.
For the large-scale preparation of transiently transfected cryobanks, Lipofectamine 2000 can cause handling difficulties as the transfection is carried out on adherent cells, and our investigations into high-density suspension protocols resulted in undesired cytotoxic effects (data not shown). Therefore, we found that moving to PEI as the transfection reagent was beneficial. It is more cost-effective, and the transfections can be carried out with the cells in suspension.
In addition to looking at the use of cryopreserved cells, a c-Myc tag was introduced to various DNA constructs and transiently transfected into the chosen cell lines and subsequently cryopreserved. Initial tagging used the monoclonal anti-c-Myc antibody 9E10, which is available commercially but was produced in-house for this investigation. The 9E10 antibody was raised to the synthetic peptide AEEQKLISEEDLL, corresponding to C-terminal amino acids 408–420 of human c-Myc. Although there is a small difference between the c-Myc tag and the epitope used to raise the antibody, we have found that 9E10 does not effectively recognize some tagged kinases. Another monoclonal anti-c-Myc antibody 9B11, raised to the smaller epitope EQKLISEEDL corresponding to C-terminal amino acids 410–419 of human c-Myc, is available from Cell Signaling Technology. This antibody recognizes all tagged kinases we have tried to date (>10), in most cases giving a better signal than 9E10 (data not shown), and so has replaced the in-house 9E10. Using the c-Myc antibody 9B11 to capture this tag meant that various targets can be processed using the same method with minimal additional reagents required.
By implementing this c-Myc tag, it was then possible to design a generic ELISA method that would measure the phosphorylation of all four targets without having target-specific total capture antibodies (which may not be readily available for all targets). To investigate the use of a generic process, the incubation times, reagents, and equipment used during the process were all standardized. For each target, a different primary (phospho or total) antibody was required, but the secondary antibodies were more standardized as either anti-rabbit (1/4000 dilution) or streptavidin-HRP (1/4000 dilution) was used. When assessing these process improvements, additional validation work was also carried out to make sure each of these targets was run using the same semi-automated processes (i.e., all dispensing was done using a WellMate, and all washes were carried out using a PW384 Platewasher). The work carried out to investigate the feasibility of moving from an overnight incubation of lysates to a 2-h incubation of lysates showed that there was little impact on data by reducing the lysate incubations. This reduction in incubation times does, however, significantly affect the time required to run a cell screen as it reduces the assay running time from 3 days to 2 days. This also means that there is faster turnaround of data from running a cell screen. In addition, the lysate volumes were increased from 25 to 40 µL to allow for additional end points to be measured using the same cell lysates. For example, a total end point of each target was also measured (data not shown) in addition to the phospho end point; the only change in process would be the different primary antibody added. The successful utilization of a standardized process means that development timelines of any new assay can be greatly reduced, as any further targets can be investigated using this method first and then the assay process optimized to suit that particular assay. This should positively affect the workload required for validation of any new assay as we no longer need to use a “one factor at a time” strategy for assay development.
Successfully optimizing the way cellular screens can be processed allows for more efficient/beneficial approaches to be fully investigated (i.e., several assays with different end points being run as one batched assay performed by one scientist). In addition, having a standardized process for running multiple ELISAs can make cell-based assays more suitable for fully automated platforms, as shown by Macmillan et al 12 in part II.
Footnotes
Acknowledgements
We thank Graham Sproat for preparing all DNA constructs. The involvement of many staff at AstraZeneca in c-Myc cellular assays has been invaluable, in particular Martina Fitzek and Jonathan Wrigley for helpful discussions in preparation of this article.