
Abstract
In the present investigation, the authors have performed an in silico–based analysis on a series of arylthiophene derivatives for the determination of their structural features responsible for farnesyltransferase (FTase) inhibitory activity, hERG blocking activity, and toxicity by quantitative structure–activity relationship and pharmacophore analysis techniques. The statistically significant models derived through multiple linear regression analysis were validated by different validation methods. The applicability of the descriptors contributed in the selected models show that the polar and polarizable properties on the van der Waals (vdW) surface area of the molecules are important for the FTase inhibitory and hERG blocking activities, while being detrimental for the toxicity of the molecules. It is interesting to note that the topological properties, molecular flexibility, and connectivity of the molecules are positively correlated to all the activities (FTase inhibition, hERG blocking, and toxicity). This implies that the flexibility of the molecules is the common feature for interaction in all targets, whereas the presence of polar groups on the molecular surface (vdW) is a determinant for the favorable (FTase inhibition) or unwanted effect (hERG blocking and toxicity) of the molecules. The pharmacophore analysis of the molecules demonstrated that the aromatic/hydrophobicity and polarizability features are important pharmacophore contours favorable for these activities.
Introduction
C
Related to ongoing and continuing research in our laboratory, our research group has been directly involved for the past 8 years in the study of the FTase enzyme, focusing mainly on the catalytic mechanism (quantum mechanical studies) of this important enzyme and the development of novel FTase inhibitors.7–11 The aim of this present investigation is to analyze the structural features (physicochemical properties) responsible for FTase inhibitory activity, as well as human ether-a-go-go-related gene (hERG) blocking activity (predicted) and toxicity (predicted) of a series of arylthiophene derivatives. 12 A quantitative structure–activity relationship (QSAR) is one of the possible techniques to study the correlation between structural features of a set of compounds needed for the interaction with its targets and their biological activities.13,14 The literature analyses show that no QSAR report has been published on this series of compounds.
hERG encodes the major protein underlying the rapid delayed rectifier K+ current in the heart. The blockade of this hERG channel has been associated with the acquired long QT syndrome (LQTS), causing ventricular tachyarrhythmia and sudden cardiac death. Nowadays, this is one of the main problems in drug design along with inappropriate ADME properties, and scientists must consider these issues while designing and developing novel bioactive moieties. 15
Hence, in the present investigation, computation-based approaches such as QSAR and pharmacophore analysis were performed on these arylthiophene derivatives to determine the structural features responsible for FTase inhibitory activity, hERG blocking activity, and toxicity. The present series has only limited compounds for the analysis, but the studies in this series will provide some useful information on the structural features responsible for FTase inhibitory activity and other toxic effects of these compounds.
Materials and Methods
A series of arylthiophene derivatives exhibiting FTase inhibitory activity was considered for the present QSAR study 12 ( Table 1 ). Compounds with defined FTase inhibitory activity were considered only for the correlation analysis. The FTase inhibitory activity of the molecules was calculated as –logIC50 or pIC50, to relate the quantitative correlation between the free energy changes and the structural features of the molecules.
Structure and FTase Inhibitory Activity of Arylthiophene Derivatives
The semi-empirical MOPAC program and the quantum semi-empirical method Austin Model 1 (AM1) with 0.05 RMS gradients of the Molecular Operating Environment (MOE) (Chemical Computing Group, Inc., Montreal, Canada) software were used to optimize the structure of the molecules and to calculate the physicochemical descriptors for the molecules. A large number of theoretical molecular descriptors (2D and 3D) are available in the package to define the structural properties of molecules explicitly, and the QuaSAR module of the MOE software was used for descriptor calculation. 16 The hERG blocking activity and toxicity (LD50) values of all the compounds in the series were predicted using q-hERG and q-Tox software, respectively (Quantum Pharmaceuticals, Moscow, Russia).
To quantify the correlation between the biological activities (FTase, hERG, and LD50) and the physicochemical descriptors, multiple linear regression (MLR) analysis was performed using Statistica 8.0 software (StatSoft, Inc., Tulsa, OK). To reduce redundant and useless information, descriptors that possessed zero correlation with the dependent variable as well as descriptors that showed an intercorrelation above 0.5 were discarded from the analysis. The stepwise MLR analysis was performed to refine the models by determining the relative importance of each variable and its statistical significance. The upper limit of the rule of thumb (six cases [compounds] per variable) was adopted in the analysis to reduce overfitting of the descriptors. 17
The significant QSAR models derived from MLR analysis were selected for further validation studies by taking into account of high correlation coefficients (R or R2), Ftest and ttest values, and the significance of the descriptors included in the model building (variance inflation factor [VIF], Durbin-Watson [DW], and beta coefficients). The selected MLR models were validated to examine the self-consistency between them, which implies a quantitative assessment of the model’s robustness and its predictive power.
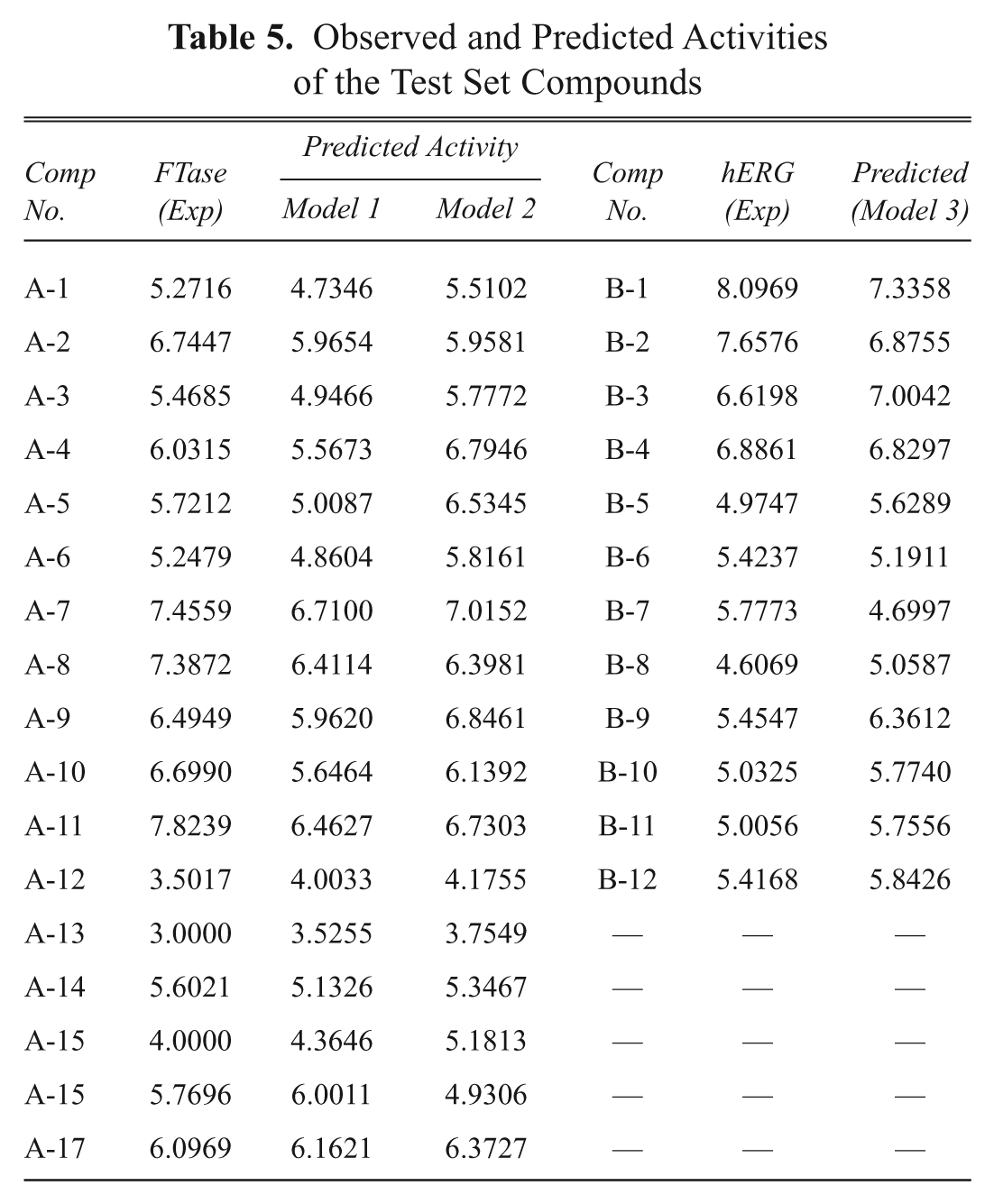
The selected MLR models were validated by leave one out (LOO), leave many out (LMO), Y-randomization, test set (external), and bootstrapping (BS) validation techniques.18,19 LMO was carried out by dividing the training set compounds into groups (blocks) of N compounds. In this study, each block had three compounds (six blocks) for FTase inhibitory activity and five compounds (four blocks) for hERG blocking and LD50 activities. The activities of the compounds were predicted from their respective MLR models (LOO and LMO), and statistical parameters were calculated to examine their predictive capacity. The Y-randomization test ensured the robustness of the QSAR model. This test was done by randomly shuffling the dependent variable vector and using the original independent variable matrix to develop a new QSAR model (20 trials were made for this analysis), and the R2 values were calculated. The number of compounds in this data set was limited, and hence we used a new data set of compounds (FTase inhibitory and hERG blocking activities) as a test set to predict their activities and validate the developed models (structures and activities provided in
The bootstrapping validation was carried out by randomly splitting the data set several times (five times) as training and test sets, and the MLR models developed with the training set (excluded test set) were used to predict the activities of the test set compounds. The predictive ability of the models was also quantified in terms of their corresponding
The pharmacophore analysis of the data set was carried out using MOE software. The conformers of the data set were developed by a stochastic search with the following computational parameters (maximum number of conformers generated to 250, superpose RMSD to 0.15, and the fragment strain limit of 4 kcal/mol). A pharmacophore search was performed on the conformers, using the aligned structure of the compounds in the series as pharmacophore query with aromatic/hydrophobic, hyd/acceptor/donor, acc, and hyd as pharmacophore contours.
Results
QSAR analysis of arylthiophene derivatives was carried out to interpret the structural features responsible for FTase inhibitory activity, hERG blocking activity, and toxicity (LD50). Multiple linear regression analysis was performed to correlate these activities and physicochemical descriptors of the molecules. The QSAR results obtained from the analysis are given below.
Model 1


Model 2
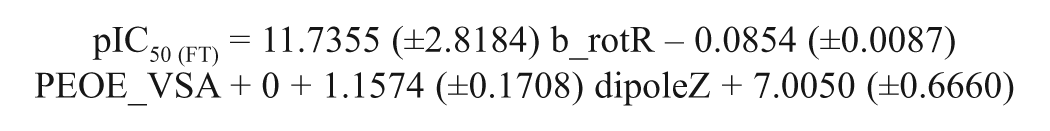
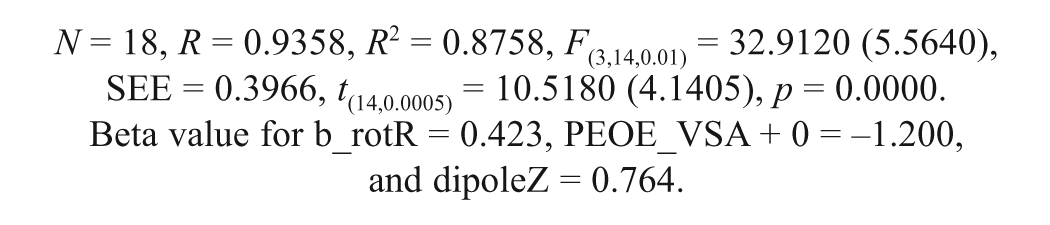
The models derived between activities such as predicted hERG blocking activity and toxicity and the physicochemical descriptors are provided as models 3 and 4, respectively. The models derived for both activities are triparametric and were developed with 21 compounds.
Model 3
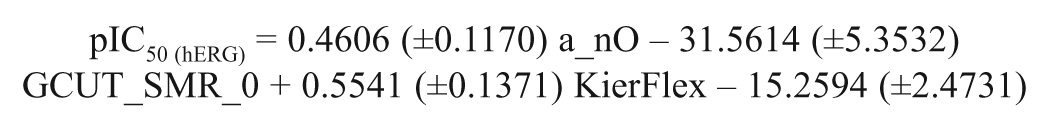

Model 4

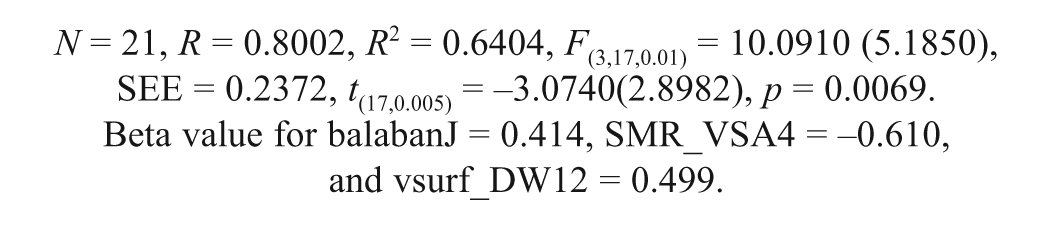
The statistically significant models (1–4) derived from the MLR analysis were validated by different validation methods to examine the self-consistency, robustness, and predictive power of the models. The results derived from the various validation methods are provided in Table 2 .
Summary of the Validation Parameters
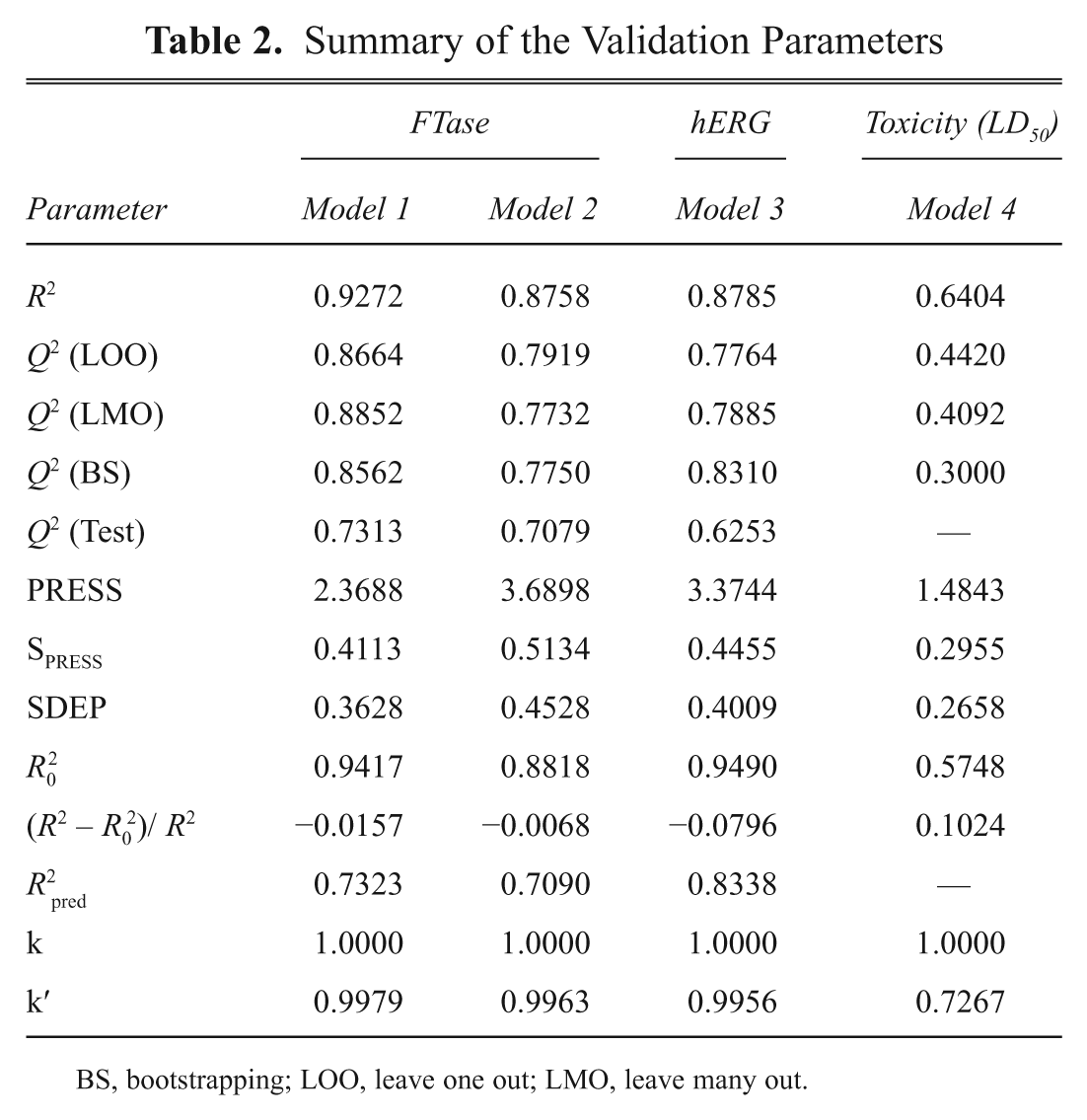
BS, bootstrapping; LOO, leave one out; LMO, leave many out.
Discussion
The triparametric QSAR models 1 and 2 are correlated between the FTase inhibitory activity and the different type of physicochemical descriptors of the molecules. The squared correlation coefficient (R2) for models 1 and 2 are 0.9272 and 0.8758, respectively, which suggests that the models possess significant fit for activity prediction and provide information that the observed activity has >87% variation.
The Ftest value indicates that the regression relations are not a chance fit but are a significant occurrence. The values within the parentheses that follow the calculated Ftest values are the tabulated values at 99% significance. t is the Student ttest, and the values within parentheses after the calculated values are the tabulated ttest values at a 0.0005 confidence level. The Ftest and ttest values have a large margin of difference from their respective tabulated values. This shows that the models are statistically significant for further validation, justifying their application to the activity prediction study.
In addition to models developed against the FTase inhibitory activity (models 1 and 2), model 3 also has significant statistical parameters, such as R2, Ftest, and ttest values. Model 3 has a correlation coefficient (R2) of 0.8785, and the Ftest and ttest values are 99% and 99.5% significant, whereas model 4 is 99% and 95% significant for Ftest and ttest values, respectively. However, model 4 has a comparatively lower correlation coefficient value (0.6404) than the previous models (1–3).
The QSAR models developed against FTase inhibitory activity show low SPRESS (0.4113 for model 1 and 0.5134 for model 2) and SDEP (0.3624 for model 1 and 0.4528 for model 2) values, revealing that the models provided small residual values between observed and predicted activities. The models constructed against the hERG blocking activity and the toxicity of the compounds show comparatively better SPRESS and SDEP values than the models derived from FTase inhibitory activity. For models 3 and 4, the latter has small values, such as SPRESS (0.2955) and SDEP (0.2658), whereas the first exhibits values around 0.4. These values reveal that the constructed models have significant predictive capacity with small predictive errors. Another parameter used to find the residual error between the observed and the predicted activity of the models is PRESS. In this analysis, models 1 to 3 give PRESS values between 2.4 and 3.9, whereas model 4 provides a value of 1.4843, which shows that the residual values for the activities of the models are smaller. The Q2 values are important parameters regarding the predictive capacity of the models because they consider the residual values of the observed activity and its average values.
A high Q2 (e.g., Q2 > 0.5) is one indicator that the model is significantly predictive.18,19,22 The cross-validated correlation coefficients (
The predictabilities of the models are also confirmed by additional validation parameters
Observed and Predicted FTase Inhibitory Activity Obtained from Different Validation Methods
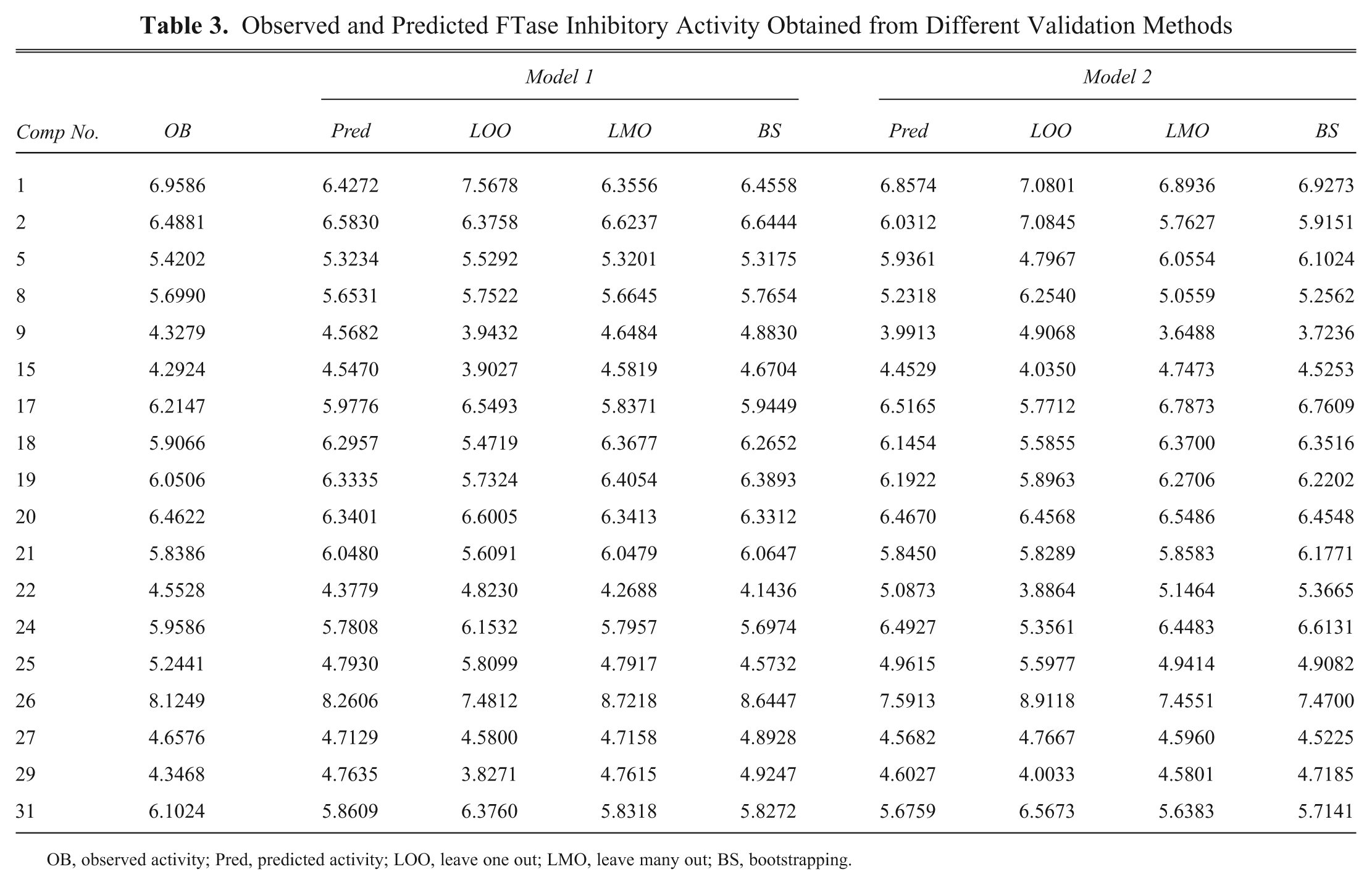
OB, observed activity; Pred, predicted activity; LOO, leave one out; LMO, leave many out; BS, bootstrapping.
Observed and Predicted hERG Inhibitory Activity and Toxicity Obtained from Different Validation Methods
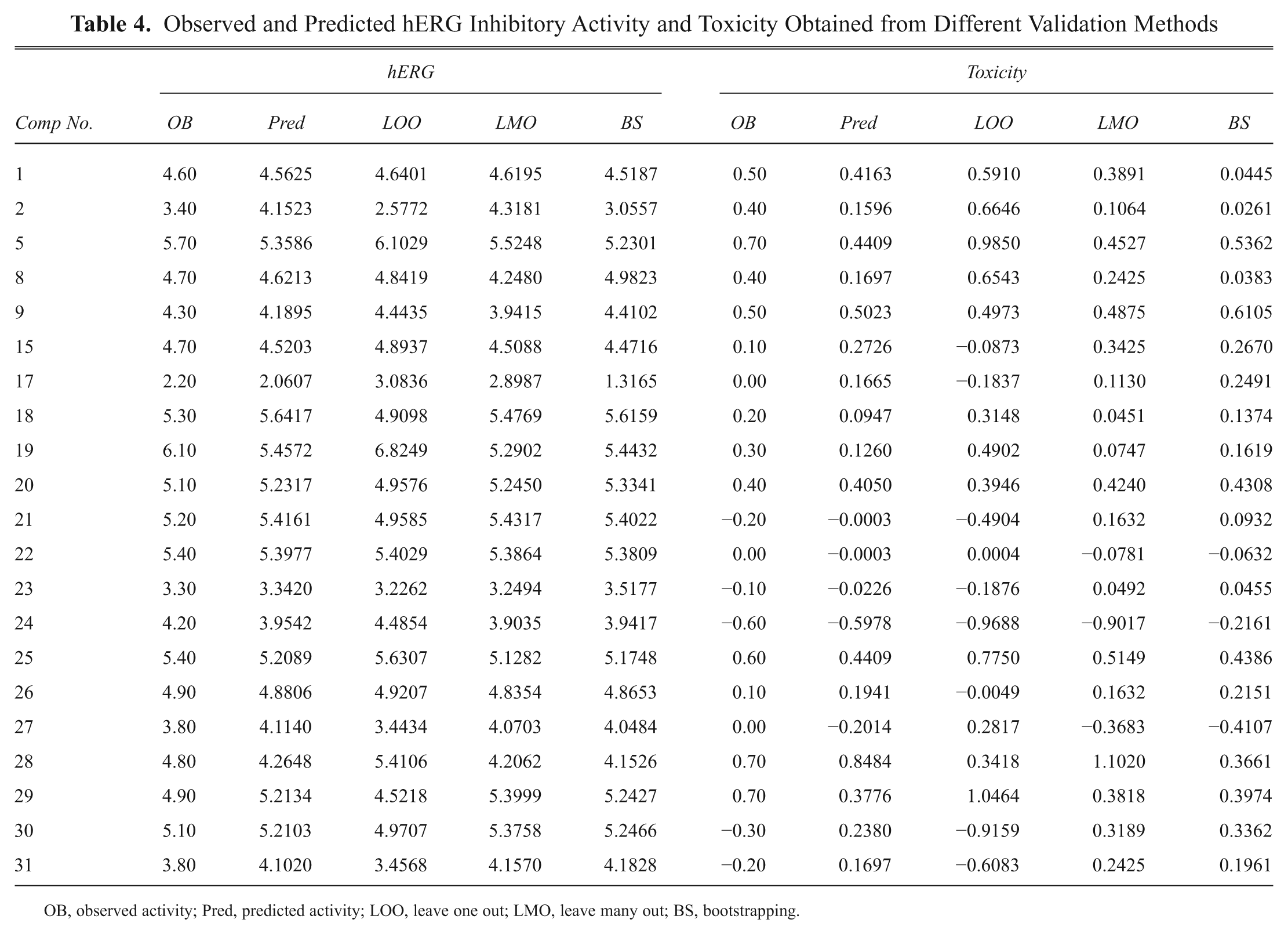
OB, observed activity; Pred, predicted activity; LOO, leave one out; LMO, leave many out; BS, bootstrapping.
Observed and Predicted Activities of the Test Set Compounds
The models were further validated by applying the Y-randomization test. The R2 values obtained from 20 trials based on permuted data are shown in Figure 1 . The R2 values of the original model were higher than any of the trials using permuted data. Hence, the models are statistically significant and robust.
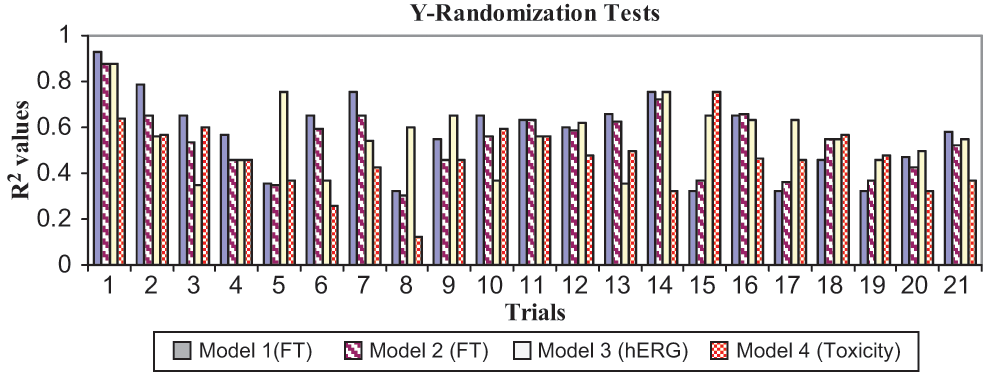
Y-randomization test result obtained from the selected models.
The validation results obtained from various validation studies reveal that models 1 to 3 are statistically significant and have predicted the activities with small prediction errors (residual errors). Model 4 (toxicity) does not provide significant validation results as compared to the other models (1–3), but this model has been considered because of the nonavailability of any other better models for toxicity.
The stability, reliability, and robustness of any models also depend on the multicollinearity and the autocorrelation of the models and the descriptors contributed to the models, respectively. Multicollinearity is a statistical phenomenon used in multiple regression models in which two or more predictor descriptors are highly correlated. Multicollinearity does not reduce the predictive power or reliability of the model as a whole; it only affects calculations regarding individual predictors.
To confirm the absence of multicollinearity, VIF values were calculated, with a VIF value greater than 10 an indication of potential multicollinearity problems. In these models, the VIF values are less than 1.6, showing that the descriptors in the selected models are free from multicollinearity ( Table 6 )—that is, none of the independent variables in the models are collinear with other independent variables in the models.11,23 It is interesting to note that model 4 provides a VIF value of 1, which is better than the value of the other models.
Variance Inflation Factor (VIF) and Durbin-Watson (DW) Values for the Significant Models
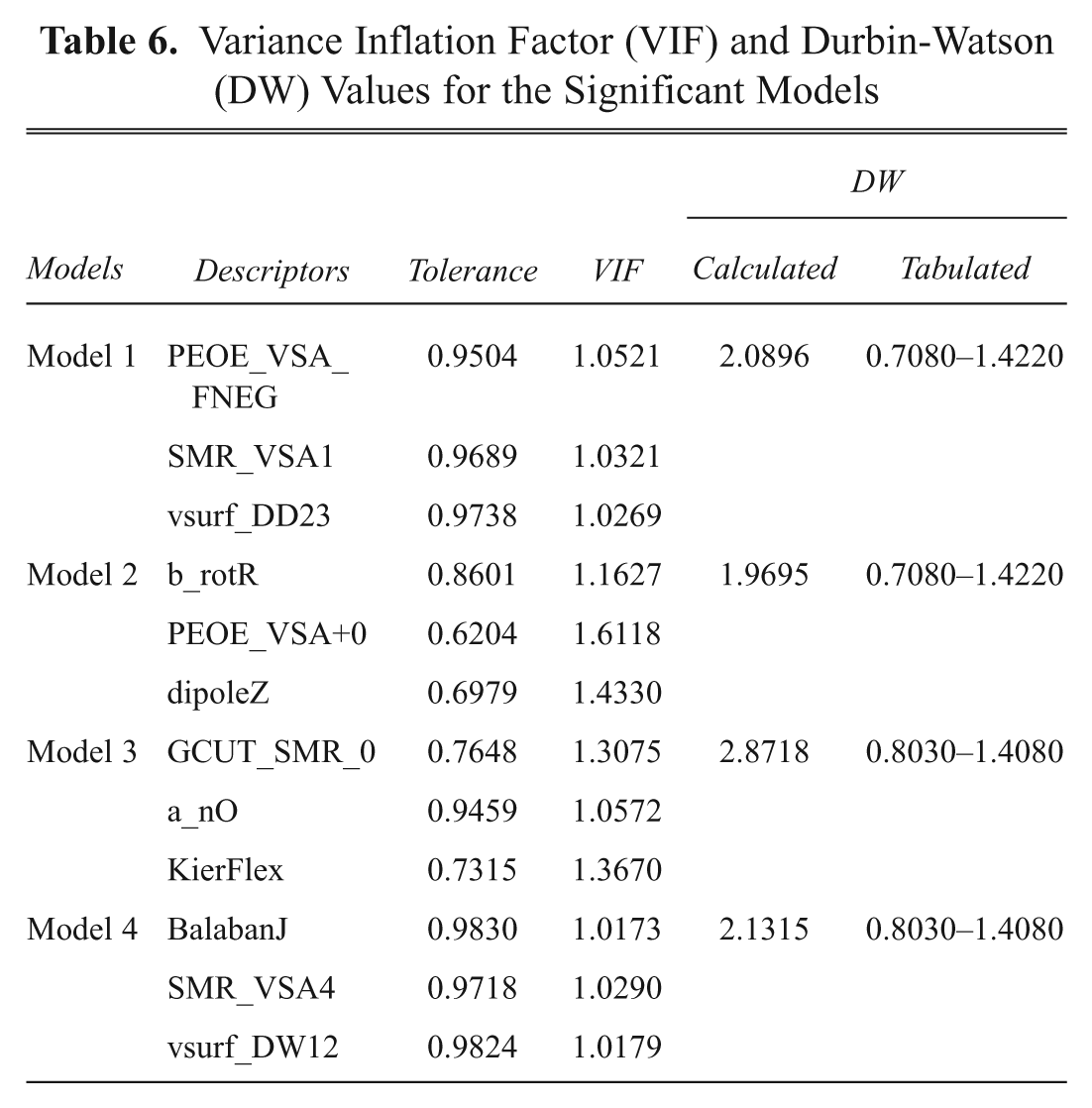
A DW test was employed to check the serial correlation of residuals (correlation of adjacent residuals). The DW statistic is useful for evaluating the presence or absence of a serial correlation of residuals, or the regression models assume that the error deviations are uncorrelated. If the DW statistic is substantially <2, there is evidence of a positive serial correlation, and a value toward 4 indicates negative autocorrelation.11,23,24
The tabulated upper and lower bound values of a DW test were considered to test the hypothesis of zero autocorrelation against the positive and negative autocorrelations. In the present study, the DW values of all models except model 3 are closer to 2, which show that the values are above the positive autocorrelation and below the negative autocorrelation of the tabulated upper and lower bound values at a 5% significance level ( Table 6 ). Model 3, at a 1% significance level for its corresponding tabulated values, does not have any serial autocorrelation. Small values of the DW statistic indicate the presence of autocorrelation, but a value less than 0.8 usually indicates that autocorrelation is likely.23,24 In the present analysis, the DW values of the models >1 show that the selected models are free from autocorrelation.
The VIF and DW values of the models show that the selected models are significant, and model 4 is totally free from multicollinearity and autocorrelation. However, this model has less significant validation parameters (cross-validated coefficients), and it may also be considered a significant model to explain the structural features of the molecules responsible for the toxicity of the compounds. These statistical analyses confirm that the selected training set models (1–4) are reliable and robust.
Applicability of the descriptors
Model 1 has been developed with the following descriptors: partial charge (PEOE_VSA_FNEG), subdivided surface area (SMR_VSA1), and surface area, volume, and shape (vsurf_DD23). PEOE_VSA_FNEG defines the fractional negative charge on the van der Waals (vdW) surface area of the molecules.16,25,26 The partial equalization of orbital electronegativities (PEOE) is a method of calculating atomic partial charges, in which the charge is transferred between bonded atoms until equilibrium. 27 This model shows that the fractional negative charge on the vdW surface of the molecule is important for FTase inhibitory activity. It reveals that some positive charged groups must be present in the active site of the FTase enzyme for better activity.
SMR_VSA1 is defined as the sum of vi for over all atoms i. pi denotes the contribution to molar refractivity for atom i as calculated in the SMR descriptor, calculated in a specified range, from 0.11 to 0.26. This descriptor reflects polarizability and the atomic contribution to molar refractivity. 16 The positively signed coefficient of this descriptor suggests that the polarizability on the vdW surface area of the molecule is favorable for FTase inhibitory activity, which reveals that the FTase enzyme should have some polarizable groups in their active site for better interaction.
The v_surf descriptors depend on the structural connectivity and conformation (dimensions are measured in Å) of the molecules. 16 The vsurf_DD23 descriptor signifies the contact distances of vsurf_DDmin, representing the distances, for the OH2 and DRY probes, between the best three local minima of interaction energy when the probes interact with a target molecule. The coefficient in this descriptor showed a negative sign, suggesting that the distance between the hydrophobic and hydrophilic region in the molecule and the target should be higher when the probes interact with the target and the should have minimum energy for better inhibitory activity.
Model 2 was constructed taking into account the FTase inhibitory activity and the physicochemical descriptors of the molecules, such as the partial charge (PEOE_VSA+0), the atom count and bond count (b_1rotR), and the conformation-dependent charge (dipoleZ) descriptors. The partial-charge descriptor (PEOE_VSA+0) provides the partial charge of an atom (qi) on the vdW surface area (Å2) of an atom (vi). This value has a range of qi (partial charge of an atom i) between 0.00 and 0.05. This PEOE_VSA+0 descriptor uses the PEOE method for the partial-charge calculation on the vdW surface area. The descriptor PEOE_VSA+0 has a negative coefficient in the model, suggesting that the positively charged groups present on the vdW surface area of the molecule are detrimental for activity. It reveals that the active site of the enzyme might have some positively charged groups for interaction with the molecule, which is in agreement with the result of the earlier model 1.
The b_1rotR describes the fraction of rotatable single bonds, and a bond is rotatable if it is not in a ring.16,28 The positive sign of the regression coefficient of this descriptor reveals that the presence of rotatable single bonds in the molecules is favorable for FTase inhibitory activity. It shows that the molecules can have more single bonds when they are in an acyclic structure, and this suggests that the compounds exhibiting a flexible acyclic structure can have significant FTase inhibitory activity.
The conformation-dependent charged descriptor (dipoleZ) is the dipole moment calculated from the partial charges (spatial separation of positive and negative charges) of the molecule at Z coordinates. The dipole moment descriptor is an electronic property that indicates the response of a molecule to an electrostatic field. The dipole moment of the molecule has been correlated to long-range ligand receptor recognition and subsequent binding. 14 The positive sign on the regression coefficient of the descriptor reveals that the dipole moment of the molecules at Z coordinates is favorable for binding to the FTase enzyme. It confirms that the active site of the enzyme may have some polar groups for interaction with the molecules.
These models (1 and 2) reveal that the partial-charge, vdW surface area, and molecular flexibility descriptors contribute to inhibitory activity prediction. The obtained results were compared with the active site characteristics of the X-ray crystallographic structures available in the protein data bank. In fact, the two available X-ray crystallographic structures along with benzofuran molecules (PDB codes 2ZIR and 2ZIS) show the existence of particularly short Zn-N bond lengths between benzofuran derivatives and the catalytically relevant Zn(II) metal ion in the FTase active site. 29
The important role of this amino acid residue (Lys164α, Arg291β, and Lys294β) for molecule binding to the FTase active site has been previously demonstrated in structural and mutagenesis studies 30 and was the subject of particular attention in some of our molecular dynamics studies, 9 suggesting that against natural peptidic CAAX substrates, this residue establishes an important interaction with the negatively charged terminal carboxylate group and makes a very important contribution to enzyme substrate affinity. In addition to this amino acid residue, several other positively charged amino acid residues have been previously implicated in substrate/inhibitor binding.29,30 This suggests that the negatively charged groups on the vdW surface of the molecule are important for interaction with the Zn2+ ion and other positively charged amino acid residues in the active site.
The correlations between hERG blocking activity (predicted) and physicochemical descriptors show that the distance and adjacency matrix descriptor (GCUT_SMR_0), the Kier and Hall connectivity indices (KierFlex), and the atom and bond counts (a_nO) descriptors contribute to hERG blocking activity prediction. The GCUT descriptors are calculated from the eigenvalues of a modified graph distance adjacency matrix.21,22 The descriptor GCUT_SMR_0 contributed negatively to the model, and the applicability of the SMR descriptors is same as that of other SMR descriptors present in earlier models. The negative contribution of the descriptor reveals that the smaller polarizability on the vdW surface of the molecules is detrimental for activity.
The topological descriptor KierFlex14,15,28 encodes the structural properties that restrict a molecule from being “infinitely flexible,” the model for which is an endless chain of C(sp3) atoms. The value of KierFlex decreases with the presence of structural features considered to prevent a molecule from attaining infinite flexibility for fewer atoms, cyclicity, branching, conjugation, and the presence of atoms with covalent radii smaller than C(sp3). The flexibility in the molecule is important for better orientation and interaction with the target protein. Another descriptor contributing to the model is a_nO, which explains that the presence of oxygen atoms in the molecules is important for activity. This shows some positively charged groups or hydrogen bonding groups in the active site for interaction with these oxygen atoms.
Model 4 was developed for toxicity (LD50) (predicted) with three physicochemical descriptors (vsurf_DW12, SMR_VSA4, and BalabanJ). The vsurf_DW12 signifies contact distances of vsurf_EWmin. The vsurf_EWmin describes the lowest hydrophilic energy representing the distances, for the OH2 and DRY probes, between the best three local minima of interaction energy when the probe interacts with a target molecule.15,23 The descriptor in this model showed positive correlation, suggesting that the interaction energies (local minima) when the hydrophilic region has contact with the target should be minimum. If the active site of the protein has hydrophobic properties, then there is less water present in the active site region, and hence the interaction energy will be low.
The subdivided surface area descriptor SMR_VSA4 describes the polarizability of the molecules on the vdW surface area of the molecules. It is calculated like the other SMR descriptors but in a specified range of 0.39 to 0.44. A negative contribution to the property in this model shows that the vdW surface area of the molecules should not have polarizable groups for the toxicity of the molecules.
BalabanJ is Balaban’s connectivity topological index, which explains the connectivity of the bonds. 16 A descriptor in this model that contributes positively to the toxicity prediction of the compounds shows that the number of edges in the molecules should be higher for better activity, which means significant connectivity of atoms in the molecules will provide toxicity.
A pharmacophore analysis on the flexialigned structure of the compounds in the series was performed, and the pharmacophoric features are shown in
Figure 2
. The flexialigned structure of the compounds shows that the hydrophobic pharmacophore contours (Aro/Hyd property) are aligned in a particular region, and the other polar pharmacophore contours (Acceptor) of the compounds are also aligned in different regions with considerable distance from the hydrophobic regions. These pharmacophore contours (polar and hydrophobic) in the flexialigned structure provide a rectangular shape with a specific distance. The distance between the Acc and Aro/Hyd contours is from 3.71 to 4.47 Å, the distance between the Aro/Hyd contours is 4.49 Å, and the distance between the acceptor contours is 6.80 Å. This reveals that the distance between the acceptor and the hydrophobic contours (Aro/Hyd) should be separated by an almost equal distance, and each acceptor contour of the molecules should be separated by a larger distance. The vdW surface properties of the flexialigned structure of the molecules also show a maximum area of the molecules surrounded by hydrophobic properties. The vdW surface properties of the reference compounds and the flexialigned structure show that the reference compounds that have significant distribution of polarity on the vdW surface of the molecules which have a better FTase inhibitory activity (
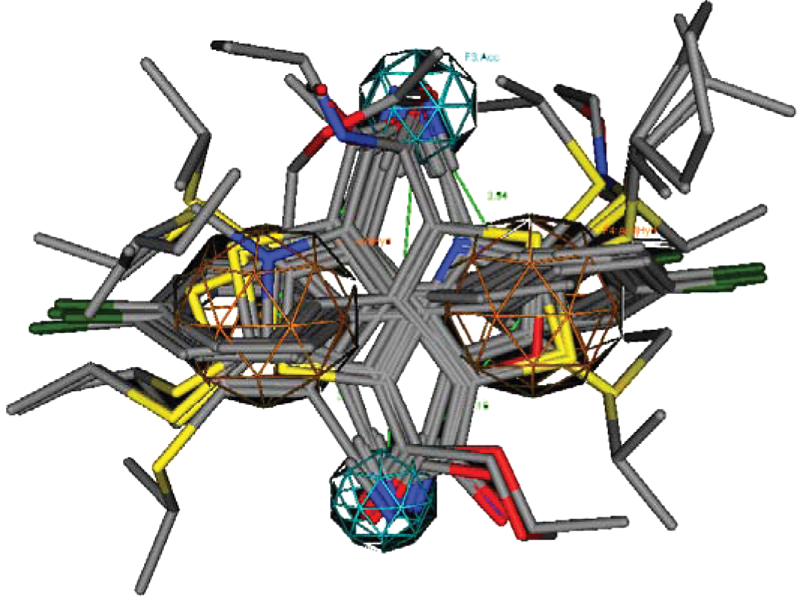
Flexialigned structure and its pharmacophore distances.
The QSAR analysis performed by the MLR method shows that a polarizability descriptor (SMR, dipole moment) and the partial-charge descriptors (PEOE) are the most important parameters for FTase inhibitory activity. Other descriptors, such as vsurf and molecular flexibility, contribute a smaller percentage than other descriptors (as per beta coefficient values). The present study confirms that the partial-charge groups (negative charge) and polarizable groups (molar refractivity) on the vdW surface area along with flexibility of the molecules are necessary for FTase inhibitory activity and hERG interaction of the molecules. This implies that the molecular flexibility of the molecules is the common property for interaction in all targets, whereas the presence of polar groups on the molecular surface determines the favorable (FTase) or unwanted effect (hERG and toxicity) of the molecules.
The SAR of the predicted hERG blocking activity with the compounds shows that the compounds with bulky hydrophobic substituents (i-Bu, s-Bu, c-Bu, N-morpholine, etc.) have hERG blocking activity at a lower concentration. Also, carboxyl substituents (CO-R) with bulky hydrophobic groups have the same kind of results as mentioned above. These results may be caused by the hydrophobic/aromatic interaction of these groups to F656 of the hERG protein. 15 The compound, which has S-CH3 in the R1 position and COOH in the R2 position, exhibits hERG blocking activity at higher concentrations. We have had the same kind of results for a lethal effect of the compounds.
These results show that the hydrophobicity of the compounds with flexibility is important for the interaction, which is in agreement with the ongoing research results of our laboratory. The predicted metabolic products of the compounds show that all the compounds undergo aromatic thioether hydrolysis and give R-SH products and may cause a toxic effect when interacting with DNA.
In conclusion, the QSAR analysis was performed with MLR analysis, and the models derived for FTase inhibitory activity, hERG blocking activity, and toxicity were validated by different validation methods. The results obtained reveal that the models are statistically significant. The study results give information on the structural relationship between FTase inhibitory and other activities (hERG and LD50). This study highlights the importance of polar groups in the molecule for interacting with the positively charged groups on the active site of the enzyme or receptor (possibly the Arg202β amino acid residue and the Zn(II) ion). This study provides guidance for better drug designs according to people’s needs for drugs that are devoid of or have fewer toxic effects (hERG and other toxic effects).
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.