
Abstract
The illumination of cellular processes in cancer has revolutionized oncology drug development leading to a shift from non-specific chemotherapy to the selective targeting of tumorigenic signal transduction pathways. Farnesyltransferase inhibitors (FTIs) target proteins needing prenylation for functioning, thus inhibiting a wide variety of molecular targets crucial for cell proliferation and survival. Tipifarnib (R115777, Zarnestra®), a potent and specific inhibitor of Farnesyltransferase, can attain strong inhibition of tumor growth in preclinical models. As a single agent, tipifarnib has demonstrated activity in several hematologic malignancies, namely acute myeloid leukemia, myelodysplastic syndrome, chronic myeloid leukemia and multiple myeloma. However, considering the complexity of the molecular aberrations implicated in the pathogenesis of hematologic neoplasms, it is rather unlikely that monotherapy with tipifarnib will serve as a stand-alone treatment approach. Indeed, improved results have been achieved by combining tipifarnib with other anticancer agents, whereas the first efforts for the identification of molecular predictors of response are reporting intriguing results. Ongoing trials are anticipated to define the exact role of tipifarnib in the treatment of hematologic malignancies.
Introduction
The recent progress in cellular and molecular biology unfolded numerous pathogenetic pathways which in turn led to a new era in drug discovery. As modern practice in oncology aims to obtain high efficacy with minimal toxicity, novel compounds targeting specific biological defects are starting to either be combined with or even replace traditional chemotherapy. In particular, the molecular complexity of the pathogenetic mechanisms in hematologic malignancies offers a plethora of distinct targets for the development of the corresponding inhibitory molecules. Most hematologic neoplasms are typically aggressive and difficult to manage, whereas the classic chemotherapeutic regimens are generally toxic, seriously compromising patients’ quality of life. Consequently, it was not surprising that drug research in Hematology has pioneered the development of targeted therapy which is gradually replacing traditional chemotherapy.
Farnesyltransferase inhibitors (FTIs) selectively inhibit the function of several proteins which possess a pivotal role in cell growth and survival.1,2 Tipifarnib (R115777, Zarnestra®) is an orally available FTI that exerts antineoplasmatic activity both in vitro and in vivo. 3 Based on its favorable toxicity profile and the wide range of cellular processes affected by tipifarnib, several investigators have conducted clinical trials in patients with acute and chronic myeloid leukemia, myelodysplastic syndrome, multiple myeloma, idiopathic myelofibrosis and non Hodgkin lymphoma.
This article reviews the current data on the role of tipifarnib in the treatment of hematologic malignancies.
Mechanism of Action
Aberrations in cell signaling pathways represent a common pattern in human cancers. 4 Constitutive activation of intracellular proteins that regulate cell survival and proliferation is implicated in the pathogenesis of most hematologic tumors, thus comprising an attractive substrate for targeted therapy. 5
Farnesyltransferase (FTase) is one of the three enzymes comprising the prenyltransferase group. 1 FTase modifies proteins posttranslationally by catalyzing the transfer of an isoprenoid lipid, namely a farnesyl moiety, to the cysteine terminal residue of substrate proteins. This process, known as farnesylation, facilitates the attachment of the farnesylated proteins to cell membranes presumably due to the hydrophobicity the farnesyl group.6,7 As most target proteins for FTase are involved in signal transduction, membrane tethering by farnesylation is crucial for their function. Tipifarnib (Zarnestra®) belongs to the group of nonpeptidomimetic FTIs and is able to exert potent and selective inhibition of FTase. Like the peptidomimetic compounds, tipifarnib imitates the cysteine terminal residue (CAAX motif) of proteins that require farnesylation, thus competing with CAAX containing peptides for FTase binding sites. 8 However, peptidomimetic FTIs displayed unacceptable toxicity in a phase I trial resulting in the cessation of further investigation of these agents. 9
The exact mechanism of action of FTIs is not completely understood. An antiproliferative effect is consistently shown in both cell lines and primary tumor cells, whereas induction of apoptosis and cell cycle arrest are less well substantiated.1,10,11 It is reported that FTIs can incite mitochondrial-triggered cell death in K-Ras transformed cell lines 12 or it can prompt apoptosis by increasing Fas-ligand expression on human cancer cell lines. 13 Nevertheless, we and others could not trace a pro-apoptotic effect of tipifarnib in CD34+ hematopoietic progenitors from MDS and AML patients.14–16
Though the development of FTIs was initially focused on the inhibition of the oncogenic Ras family of proteins, an increasingly large number of proteins are identified as substrates for FTase. Beyond Ras, compelling data support a role for RhoB, nuclear lamin and centromere-associated proteins17,18 and transcriptional modulation of transforming growth factor (TGF) 19 and vascular endothelial growth factor (VEGF) 20 in the anti-tumor effects of FTIs (Fig. 1). Moreover, as mentioned above, tipifarnib may act independently of FTase inhibition, by repressing Pgp-mediated drug efflux. 21
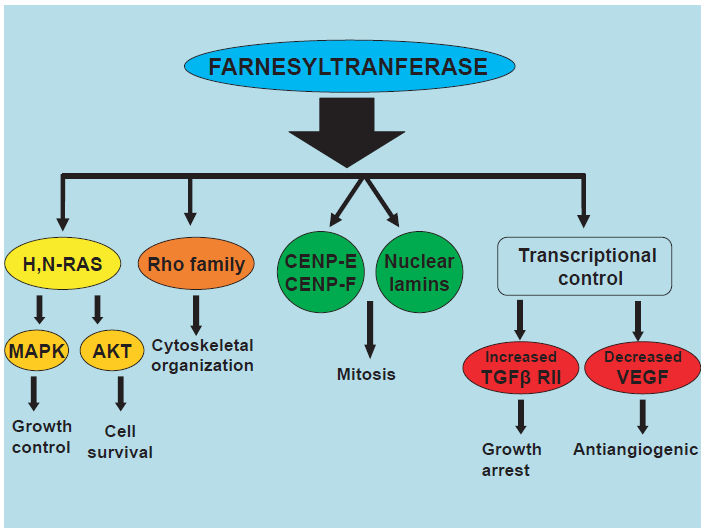
Reported targets of farnesyltransferase inhibitors.
RAS
The Ras family of oncoproteins acts as key modulators of cell signaling pathways and its members are involved in cellular proliferation, survival and differentiation.22,23 Attachment of Ras proteins to the cell membrane, a critical posttranslational step that is catalyzed by farnesyltransferase (FTase), is essential in order to become fully functionally active. 24
The impressively high frequency of Ras mutations in human neoplasms is linked with the induction of excessive mitogenesis and the promotion of tumor growth. 25 As regards hematologic malignancies, activating mutations of N-Ras and less often of K-ras have been reported in about 10%-40% of MDS26,27 and 15%-25% of AML patients28,29 and have been shown to contribute to the leukemic transformation in the former, although this was deduced indirectly by the correlation of the emergence of new Ras mutations with MDS progression. Other hematologic tumors bearing Ras mutations include multiple myeloma30,31 and acute lymphoblastic leukemia.32,33 Interestingly, inhibition of Ras by tipifarnib does not seem to directly relate to the antitumor efficacy of the latter, as in several clinical trials tipifarnib exhibited clinical activity in patients without Ras mutations, while the degree of FTase inhibition was unrelated to the clinical response.34–36 In addition, preclinical models further uncouple FTI action from RAS alteration by showing that inhibition of the malignant growth in N-Ras and K-Ras transformed human cell lines is Ras-independent, as the latter can still retain its membrane-bound active form in FTI-treated cells bypassing the need for farnesylation via geranylgeranylation. 37
RAS Downstream Pathways
At least two Ras-mediated signaling cascades have been shown to be targeted by FTIs. The first is the Raf-MEK-MAPK pathway, which holds a central role in cell proliferation and survival. Raf protein binds to Ras and then initiates a sequential phosphorylation of various kinases eventuating in the activation of mitogen-activated protein kinase (MAPK).38–40 In untreated AML patients, constitutive activation of MAPK in bone marrow was inversely related to survival,41,42 while in multiple myeloma cell lines MAPK activation induced the propagation of tumor cells that was reversed by pharmacological blocking of MAPK.43,44 FTIs can inhibit MAPK in H-Ras-transformed NIH 3T3 cells, but this effect correlated strongly with the extent of Ras inhibition suggesting that MAPK itself may not be directly targeted by FTIs.45,46
The second Ras downstream cascade is the phosphatidylinositol-3 kinase (PI3K)/AKT pathway, which, among its plethora of functions, is also critical for cell proliferation and survival.47,48 The importance of the (PI3K)/AKT pathway in malignant hematopoiesis is supported by numerous studies addressing its expression and function in CML, 49 AML 50 and NHL. 51 Inhibition of the above pathway by FTIs can be dependent or independent of Ras inhibition and can enhance apoptosis in human cancer cells, 52 although another study conversely suggested that activation of the (PI3K)/AKT pathway might render tumor cells resistant to FTIs. 53
RhoB
Rho proteins are small GTP-binding proteins which also belong to the Ras family and regulate intracellular actin dynamics and trafficking of growth-factor receptors. 54 Prenylation occurs in Rho members, which are also is increased in human malignancies, 55 thus constituting relevant targets for FTIs. The effect of FTIs in the formation of actin stress fibbers in normal cells propelled the further investigation of the Rho proteins; 56 subsequently, RhoB was identified as an essential target of FTIs. 57 Ras transformation depends highly on RhoB activity, as inhibitory mutants of the latter suppress the H-Ras-induced transformation of cell lines, 58 whereas overexpression of N-myristoylated RhoB in Ras-transformed cells ablated the antiproliferative effect of FTIs. 57 Blocking RhoB farnesylation with FTIs leads to the accumulation of the geranylgeranylated form of RhoB and subsequent growth inhibition. 59 Notably, RhoB knockouts, though highly susceptible to carcinogen-induced tumorigenesis, display normal hematopoietic differentiation, 60 whereas both in vitro 59 and ex vivo14–16 assays also showed that normal cells are significantly less susceptible to FTIs compared to malignant cells. Accordingly, RhoB is currently considered as one the most important targets of FTIs, even though opposed findings are reported by groups showing that both farnesylated and geranylgeranylated forms of RhoB are upregulated by FTIs and can inhibit tumor progression in human cancer cell lines and nude mice.61,62
Nuclear Lamins
The nuclear lamina is a scaffolding network inside the nucleus and just beneath the inner nuclear membrane. 63 Besides its role in mechanically supporting the nucleus, the nuclear lamina participates in numerous cellular events, such as cell cycle regulation, DNA replication, cell differentiation and apoptosis. 64 Farnesylation of the nuclear lamina proteins, lamin A and lamin B, is critical for their assembly to the nuclear envelope and can be inhibited by FTIs,17,65–67 as was further confirmed recently in a mouse model of progeria in which tipifarnib prevented both the onset and the late progression of cardiovascular disease by inhibiting the farnesylation of the mutant form of lamin A (progerin). 68
CENPs
FTIs also target the kinetochore-binding centromeric proteins (CENPs) E and F which are responsible for the formation of the bipolar spindle during mitosis.69,70 Even though FTI-treated cells show normal localization of CENPs to the kinetochores, the association of the above proteins with the microtubules is impaired, culminating in cell cycle arrest in prometaphase. 71 Interestingly, FTIs exhibit enhanced antitumor activity when combined with antimicrotubule agents thus further supporting the aforementioned antineoplastic mechanism.72,73
Transcriptional Control of Transforming Growth Factor (TGF) and Vascular Endothelial Growth Factor (VEGF)
Decreased expression of type II TGFβ receptor (TGFβ RII) is a ras-dependent mechanism of cancer escape that leads to dysregulated TGF signaling and uncontrolled tumor growth. 74 FTIs can enhance TGFβ RII expression 75 and restore the antitumor effect of TGFβ by inhibiting DNA methyltransferase 1 (DNMT1) as was shown in pancreatic cancer cell lines. 19
Antitumor activity of FTIs in Ras-transformed cells 13 and mouse xenograft models 76 has also been associated with reductions in tumor vascularity and VEGF levels. However, although experimental data suggest that FTIs induce a direct insult to endothelium 77 and VEGF inhibition can occur in the absence of Ras mutations, 78 clinical studies report discordant conclusions regarding VEGF levels in FTI-treated patients.36,79
Metabolism and Pharmacokinetic Profile
Most pharmacokinetic data are taken from phase I and II trials in patients with advanced solid tumors, but there are also studies in healthy subjects. 80 Despite the multitude of different schedules used in the above studies, tipifarnib pharmacokinetics appear to be linear in doses up to 600 mg/d, thus allowing the assessment of its oral bioavailability. 81 Tipifarnib is absorbed rapidly after oral administration and reaches peak plasma concentrations in 2-4 hours, 82 while the plasma levels follow a biphasic time curve with a half life of 2-5 hours in the early phase and a terminal half life of 16-20 hours. 83 Absorption rate is faster in healthy individuals (1.63 h-1) compared to cancer patients (0.71 h-1), who also have lower rates of systemic clearance of the drug. 84 However, both the steady-state volume of distribution and the extent of absorption did not differ between normals and cancer patients. Tipifarnib binds almost totally to plasma proteins and is thoroughly metabolized by the liver and is virtually undetectable in the urine. 67 The absolute bioavailability of tipifarnib is 26.7% and is similar among the various formulations and between healthy volunteers and cancer patients.84,85
Clinical Studies: Efficacy
AML Studies
Phase I Studies
Acute myeloid leukemia poses a serious therapeutic problem particularly in the elderly, as the current treatment strategies have poor results and are frequently accompanied by debilitating toxicity. Tipifarnib inhibited both normal and leukemic CD34+ cell proliferation in vitro, but spared the long-term culture initiating cells and allows NOD-SCID mouse reconstitution, 16 suggesting that the target cell population of tipifarnib consists of intermediate to late stage hematopoietic progenitors. Interestingly, tipifarnib did not induce significant apoptosis in leukemic cells nor cell cycle changes, 16 but it has been shown to possess potent MDR1-inhibitory properties. 21
The first phase I trial in AML was a single centre dose escalation study, in which tipifarnib was administered orally in 24 patients with relapsed (n = 9), refractory (n = 10) and poor risk (n = 6) AML. 67 The study also included 6 patients with acute lymphocytic leukemia (3 with Ph+ ALL) and 3 patients with chronic myeloid leukemia (CML) in blast crisis. Tipifarnib doses ranged from 100 mg to 1200 mg twice daily (BID) for up to 21 days and 8 out of 25 AML patients responded, including two complete remissions. FTPase inhibition was observed in doses of 300 mg BID or higher, whereas inhibition of protein farnesylation, assessed by the accumulation of prelaminin A and pre-HDJ-2, required at least 600 mg of tipifarnib BID. Notably, no RAS mutations were found in any of 34 patients, responding and non-responding, suggesting that tipifarnib acts via RAS-independent mechanisms.
The second phase I study utilized a different protocol in 30 refractory/relapsed AML patients. 86 Tipifarnib was administered one week on one week off in 28-day treatment cycles at doses ranging from 400-1600 mg BID and 3 of the patients achieved a complete response (CR) and 2 hematological improvements (HI), one platelet response and one downgrading back to CMML. All of the responding patients required doses of 800 mg BID or higher, while the complete responses were observed at 1000-1200 mg BID.
Phase II Studies
Based on the promising results of phase I trials, several phase II studies were conducted in order to evaluate the efficacy of tipifarnib either as induction or as maintenance therapy for AML. Two open-label multicentre studies were launched in Europe (13 countries) and the US. In the first study, 87 252 patients with refractory (n = 117) or relapsed (n = 135) AML received 600 mg tipifarnib BID for the first 21 days of 28-day cycles. The response rate was very low, with only 11 (4%) patients achieving a complete response, while two of them exhibited incomplete platelet recovery (CRp). Time to response was 58 and 68 days for patients with CR and CRp, respectively and the median duration of response was 78 days. However, a bone marrow response was evident in 134 (53%) patients with 19 of them having less than 5% blasts and the median survival of the 11 patients was 369 days compared to 87 days for the total patient population. The US study 88 enrolled 160 elderly (median age 74 years) patients with untreated poor risk AML who were not candidates for conventional chemotherapy. Seventy-five percent of the patients had progressed from MDS and 7 patients had high risk CMML. Tipifarnib was administered at 600 BID for 21 days with a 1 to 3 week recovery period among cycles. A response was attained in 37 patients with 22 CR (14%), 3 partial remissions (PR) and 12 hematological improvements (HI). Interestingly, complete remissions were more frequent in patients >75 years old (20%) and lasted for 7.3 (5.1-12.5) months. The median overall survival (OS) was 18 months for patients who achieved CR compared to 5.3 months in the overall patient cohort. In univariate analysis, older age, unfavourable karyotype, high blast count in the bone marrow and bad performance status were inversely correlated with OS and response rate. In addition, inhibition of HDJ-2 farnesylation was found to be a prerequisite for CR, but there was no significant difference between baseline phosphorylation levels of mitogen-activated protein kinase (MAPK) and AKT between responders and nonresponders.
Another multicentre phase II trial evaluated tipifarnib as maintenance therapy in poor risk AML patients after first CR. 89 Tipifarnib was administered at 400 BID for 21 days in 28 day cycles in 48 AML patients with median age of 63 years. A total of 16 cycles were completed in 20 (42%) of the patients, whereas 24 (50%) relapsed and were removed from the study. The median disease-free survival (DFS) was 13.5 (3.5-59+) months and 30% of the patients achieved a DFS of more than 2 years.
The results of the above studies, combined with the convenient administration of on an outpatient setting, prompted the further investigation of tipifarnib in the treatment of elderly AML patients with poor performance status.
Phase III Studies
Only one phase III trial of tipifarnib as monotherapy in AML patients has been conducted as yet. Results of this multicentre, randomized, study which have included 457 elderly, newly diagnosed, AML patients unable or unwilling to receive intensive chemotherapy have just been published. 90 Patients were randomized for age (<75 vs. >75) and PS (0-1 vs. 2) and then received either 600 mg tipifarnib BID orally for 21 days in 28 day cycles, or supportive care including hydroxyurea. Overall survival was identical in the two arms, 107d for tipifarnib versus 109d for best supportive care and the authors concluded that tipifarnib treatment did not confer a superior survival in these patients. Also, the CR rate was only 8%, thus lower than phase I and II trials, although the responses were relatively durable with a median duration of 8 months.
MDS Studies
Phase I Studies
As approximately 10%-40% of patients with MDS harbour Ras mutations it is not surprising that several investigators studied the activity of FTIs in this incurable malignancy. In particular, tipifarnib is the most extensively studied FTI in MDS. Preclinical data showed that tipifarnib exerts selective toxicity against MDS progenitors and this effect was not due to the induction of apoptosis. 14 The first dose-escalation trial included 21 patients of all MDS subtypes with a median age 66 years. 34 The starting dose was 300 mg BID orally for 21 days in 28 day cycles and was escalated by 100 mg per day until grade I toxicity occurred. The maximum tolerated dose was 400 BID and objective responses were seen in 6/30 patients (30%), including 1 CR, 2 PR and 3 HI, but only 2 of the responders had Ras mutations suggesting that tipifarnib targets signaling molecules downstream of the Ras pathway. A subsequent phase I study utilized an alternate week dosing schedule in 63 patients with advanced MDS in their majority. 91 Tipifarnib was given orally escalating from 100 mg BID to 1200 mg which represented the maximum tolerated dose. The response rate was 26% (16/63) with 3 CR and 13 HI. Notably, there was no dose-response correlation, whereas major platelet responses were seen in 11 out of 16 responders. Moreover, this study further confirmed the absence of any relationship between the response in tipifarnib and Ras mutational status, as only one responder bore a Ras mutation.
Phase II Studies
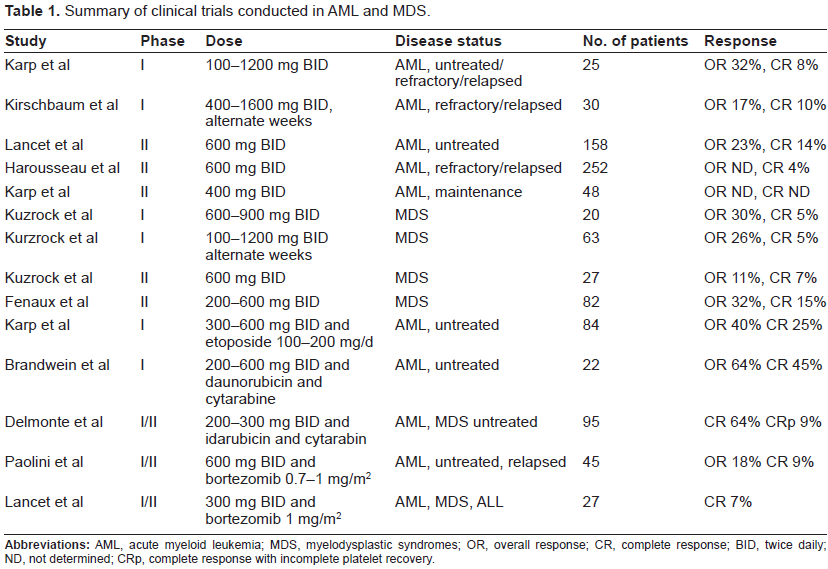
Despite the poor responses seen in phase I trials, two phase II open-label studies in MDS patients followed and are now completed. Kuzrock et al treated 27 late-stage MDS patients with median age 66 years at a starting tipifarnib dose of 600 mg BID for 28 days in 6 week cycles. 36 Only 3 responses were noted (2 CR and one PR), but full evaluation was hampered by the discontinuation of the regimen in 41% of the patients due to unacceptable toxicity. Based on these observation, a second, international study used a more tolerable regimen in 82 patients with high risk MDS. 92 Tipifarnib was administered at 300 mg BID for 21 days in 28 day cycles allowing escalation of up to 600 mg BID for patients not achieving a CR. An impressively higher response rate was observed (26/82 patients, 32%), including 12 CR (14%) and 14 HI (17%). The median OS and CR duration were 11.7 and 11.5 months, respectively. The above remarkable results were further accompanied by independence in platelet and red cell transfusion in 27% and 11% of the patients, respectively and 3 complete cytogenetic responses. A summary of tipifarnib trials in AML and MDS is shown in Table 1.
Summary of clinical trials conducted in AML and MDS.
Studies in Chronic Myeloid Leukemia and Myeloproliferative Disorders
The successful outcome of chronic myeloid leukemia (CML) with tyrosine kinase inhibitors (TKIs) was a milestone in cancer therapeutics. However, both primary and secondary resistant in TKIs may occur, whereas tolerability of the available TKIs is also a concern, particularly in elderly patients. FTIs other than tipifarnib have shown activity against imatinib-resistant CML and Ph+ ALL both in vitro and in vivo. Until today, only one study explored the efficacy of tipifarnib as monotherapy in 22 CML patients, 17 (77%) of which were resistant, refractory or intolerant in imatinib. 79 Tipifarnib was given at 600 BID for 4 weeks in 6-week cycles. Only 7 patients (6 in chronic and one in advanced phase) experienced a transient CR or PR, while 4 minor cytogenetic responses occurred (3 in chronic and one in advanced phase) and there appeared to be a relationship of high plasma levels of vascular endothelial growth factor (VEGF) with treatment response.
Tipifarnib has also been shown to be active in vitro against malignant progenitors of idiopathic myelofibrosis (IMF) and polycythemia vera (PV) patients, in clinically attainable concentrations.93,94 Interestingly, as reported in MDS, PV clonal progenitors displayed an augmented sensitivity in tipifarnib compared to their non-clonal counterparts. 94 In the aforementioned MD Anderson study, 79 2/8 patients with IMF receiving 600 mg BID obtained a notable decrease in splenomegaly and one of them became transfusion independent. Tipifarnib was also administered at 300 mg BID for 21 days in 28 day cycles in 28 IMF and 6 post-PV or post-essential thrombocythemia (post-ET) myelofibrosis patients, in a phase II trial. 95 All patients were anemic and had palpable splenomegaly. Five out of 13 (38%) of patients who required regular transfusions became transfusion independent and 11 (33%) of the patients exhibited an improvement in organomegaly, but anemia was not ameliorated in most individuals, presumably because tipifarnib myelotoxicity counterbalanced any beneficial effect of the IMF clone reduction.
Myeloma Studies
The first preclinical data on the activity of tipifarnib against multiple myeloma (MM) was demonstrated by Le Gouill et al, who found that primary myeloma plasma cells were selectively targeted by tipifarnib, which also inhibited the phosphorylation of signalling proteins involved in tumor survival and/or proliferation. 96 Since Ras mutations occur in about 30%-40% of MM patients subsequent studies addressed whether the former constitute a target for tipifarnib, but results were conflicting as induction of apoptosis was found to be Ras dependent in one study, 97 whereas two other studies indicated primarily Ras-independent mechanisms.98,99
Despite the encouraging findings in vitro, the single existing phase II trial of tipifarnib in MM showed only modest activity of this agent in 43 heavily treated patients with advanced disease. 100 No complete or partial responses were observed, but 64% of the patients receiving 300 mg BID for 21 days in 28 day cycles achieved disease stabilization, defined as a less than 50% reduction in M-component. Moreover, in agreement with most of the studies in AML and MDS, this effect was not correlated with inhibition of farnesylation or the presence of Ras mutations.
Lymphoma Studies
Refractory or primary resistant non Hodgkinlymphoma (NHL) has a grave prognosis and represents a typical example of almost invariable inefficacy of the available therapeutic armamentarium. Up to date there is limited information on the activity of tipifarnib in NHL, both in the preclinical and clinical settings. Mantle cell lymphoma (MCL) cell lines displayed significant growth inhibition and apoptosis after exposure in tipifarnib, while mice xenografted with the UPN1 MCL cell line also showed tumor stability after administration of high doses of tipifarnib. 101 Similarly, Hodgkin lymphoma (HL) cell lines were sensitive to tipifarnib which might also inhibit Pgp-mediated doxorubicin efflux in these cells. 102
In a phase II study, 42 patients with relapsed diffuse large cell (DLBCL, n = 37), follicular grade III (n = 1), or mantle cell lymphoma (n = 4) received tipifarnib at 300 mg BID for 21 days in 28 day cycles. Seven (18%) patients, all with DLBCL, obtained a PR which lasted from 4.2 to 25.1 months, but the median progression free survival for all patients was only 2 months. 103 Another small study evaluated tipifarnib treatment in 11 patients with stage IV, refractory, MCL. 104 Only one CR was observed, accompanied by an increase in the RASGRP1/APTX gene expression ratio and a decrease in AKAP13 expression. Inverse results were seen in 2 non-responding patients and the authors concluded that assessment of the above genes could predict response of MCL to tipifarnib, as was previously suggested in AML. 105 A single case of a NK-large granular lymphocyte (LGL) leukemia complicated with pulmonary artery hypertension that responded in tipifarnib, presumably via inhibition of the Ras/MAPK pathway, has also been described. 106
Combination Studies
AML/MDS
Tipifarnib primarily inhibits cell proliferation, therefore it could theoretically induce an additive effect if combined with classic cytotoxic chemotherapy.
In a phase I study, tipifarnib was administered in 84 elderly, poor risk AML patients at 300-600 mg BID for 14 or 21 days plus etoposide 100-200 mg daily on days 1-3 and 8-10. Despite their poor performance status, 21 (25%) of the patients reached a CR with a median duration of 9.8 months. Interestingly, patients in the 14-day schedule achieved better results. Moreover, as inferred by the increased histone H2AX phosphorylation and apoptosis, the combination of tipifarnib with etoposide significantly increased the magnitude of DNA damage, exclusively in patients who achieved a CR. 107
Another phase I/II study 108 evaluated the combination of tipifarnib with idarubicin and cytarabine in 95 newly diagnosed AML or high-risk MDS patients. The therapeutic schedule consisted of idarubicin 12 mg/m2 daily on days 1-3, cytarabine 1.5 g/m2 as 24 hour infusion on days 1-4 and tipifarnib 200-300 mg BID for 21 days in 28 day cycles. Patients in CR received consolidation with idarubicin, cytarabine and tipifarnib 300 mg BID for 14 days every 4-6 weeks, while maintenance therapy with tipifarnib was administered for 6 more months at doses similar to the consolidation regimen. Sixty-one patients (64%) achieved a CR, 9 (9%) a CRp and the median CR duration and OS was 72 weeks and 70 weeks, respectively, indicating that the addition of tipifarnib in the classic chemotherapy of AML did not impart an improved outcome. Moreover, the combination of tipifarnib (300 mg BID for 21 days at 6-8 weeks interval) with low-dose cytarabine in 65 elderly patients with untreated AML resulted in excess mortality, leading to the premature closure of the trial. 109
Based on the in vitro synergism against AML cell lines, 110 tipifarnib was also combined with the proteasome inhibitor bortezomib in two phase I/II trials. In the first study, 111 27 patients with advanced AML received tipifarnib at a starting dose of 300 mg/m2 and bortezomib 1 g/m2. Only two patients achieved a CR, but results are still premature and updated data are awaited. The second study 112 enrolled 45 patients with poor-risk AML who received 600 mg of tipifarnib BID for 21 days in 28 day cycles and bortezomib at 0.7 mg/m2 as weekly infusion for 3 consecutive weeks. Two patients achieved CR, one a PR and one a HI and all responses were accompanied by a higher RASGRP1/APTX ratio.
CML
The combination of tipifarnib with imatinib in imatinib-resistant CML was also assessed in two phase I trials. The first study enrolled 12 113 accelerated or blast phase CML patients. Tipifarnib was given at 200 mg or 300 mg BID together with imatinib 600 mg/day or 400 mg/day respectively. No cytogenetic and 3 complete hematologic responses occurred, one of which in a patient in blast crisis. The second study 114 included 26 CML patients in chronic phase who received tipifarnib at a starting dose of 300 mg BID for 14 days every 21 days and imatinib at 300 mg/day, both escalated at final doses of 400 mg BID and 400 mg/day, respectively. Seventeen (68%) patients achieved hematologic responses and 9 (36%) a cytogenetic response (3 complete, 4 partial and 2 minimal responses), including one patient bearing the T315I mutation (PR), but the median response duration was only 3 months.
Myeloma
As mentioned above, in vitro studies demonstrated profound synergy of tipifarnib with bortezomib against AML and myeloma via several independent pathways. One recent international, phase I, trial has been presented in the 2008 ASH annual meeting. 115 In this study, 16 patients with relapsed or refractory myeloma received bortezomib at 1.0 mg/m2 given on days 1, 4, 8, and 11 along with escalating doses of tipifarnib (100-400 mg BID) given on days 2-15 every 21 days. Stabilization of the disease was observed in 5 patients and 2 additional patients achieved a minor response.
Clinical Studies: Safety and Tolerability
Tipifarnib therapy is generally well tolerated in the majority of the patients, independently of the disease. In most phase I and II trials, the maximum tolerated dose (MTD) was determined at 1200 mg BID, while in the remaining studies MTD of tipifarnib ranged between 1000 to 1600 mg BID, depending on the dosing schedule. However, various toxicities do occur, especially when tipifarnib is administered at doses above 600 mg BID or in combination regimens and are listed below.
Myelotoxicity
Hematologic toxicity is the most common side effect of tipifarnib treatment. In a phase I study in AML, dose-limiting myelosuppression was noted in 7/8 patients at doses 600-900 BID and the nadir of neutropenia occurred on median day 16 (range 3-22), whereas no myelotoxicity was observed in lower doses. 67 Comparable findings were reported in AML patients receiving 600 mg BID tipifarnib in an alternate week schedule. 86 Myelosuppression (all grades) occurred in 60% of the patients, but only doses higher than 600 mg BID required adjustment. The above rates of myelotoxicity were further confirmed in phase II trials in AML employing analogous dosing schemes of tipifarnib. Nevertheless, cytopenias are a common finding in AML patients with advanced and/or heavily treated disease, thus discerning the cause of a further decrease in hematological values, namely drug toxicity versus disease progression, is not always feasible and should be interpreted with caution.
The same problem also applies in MDS studies, the majority of which reported identical rates of myelosuppression with the AML trials. However, the lower tipifarnib dose (300 mg BID) utilized in the most recent phase II study in MDS, 92 resulted in a much lower incidence of drug-induced neutropenia (18%), thrombocytopenia (32%) and anemia (18%). Notably, patients with IMF appear to be more sensitive to the myelotoxic effects of tipifarnib, as, even with the 300 BID schedule, 38% of the cohort developed grade 3 myelosuppression. 95
As regards combination of tipifarnib with other agents, the addition of tipifarnib in the classic chemotherapeutic regimens of AML and high-risk MDS did not significantly affect the hematologic recovery after induction chemotherapy. 116 On the contrary, the combination of tipifarnib with imatinib in chronic phase CML patients resulted in significant myelotoxicity compared to tipifarnib monotherapy and was the main reason for treatment discontinuation and patient hospitalization during the study period. 114
Non-Hematologic Toxicity
Tipifarnib can inflict a wide variety of non-hematologic toxicities. In phase I trials in AML fatigue, nausea and renal dysfunction were the most common when tipifarnib was administered in doses up to 600 mg BID. 67 However, only creatinine elevation was considered as a dose-limiting toxicity (DLT) in higher doses. Hepatic failure and neurotoxicity consisting of grade 3 ataxia and confusion were the other DLTs in these studies. In subsequent studies in AML, hypokalemia, diarrhea, skin rash and bilirubin elevation were the most frequent non-DLT.87,88
Fatigue, nausea, rash and hepatotoxicity were also the most commonly observed non-hematologic toxicities in phase I MDS studies.34,91 Dose-limiting neurotoxicity consisted of ataxia and confusion occurred, but, in contrast to AML, the main DLT was fatigue. The utilization of the 300 mg BID scheme in the most recent phase II trial 92 resulted in the complete absence of grade 4 and an impressively low rate of grade 3 non-hematologic toxicities (rash: 4%, fatigue: 2%).
As expected, the combination of tipifarnib with other compounds increased the rate and the type of non-hematologic toxicities. High-risk MDS and AML patients displayed high incidence of grade 3 and 4 diarrhea, hypokalemia, rash and hepatic dysfunction when treated with intensive chemotherapy plus tipifarnib (300 mg BID), necessitating dose reductions in 56% of the patients. 108 Additionally, grade 3 typhlitis and supraventricular tachycardia occurred in AML patients receiving cytarabine and daunorubicin together with tipifarnib at 600 mg, 117 whereas the combination of tipifarnib (300-600 BID) with etoposide induced considerable mucositis and neurotoxicity in nearly one fifth of elderly patients with AML. 107 Grade 3-4 diarrhea, fatigue and esophagitis leading to treatment discontinuation were also observed in chronic phase CML patients receiving tipifarnib at 400 BID along with imatinib 400 mg/day. 114
In contrast to the above, the combination of tipifarnib with bortezomib in both AML and MM patients111,112,115 seems to be particularly well tolerated, as only 4 AML patients to date have experienced DLT, consisting of sensory neuropathy (2), gastrointesinal toxicity (1) and fatigue (1), while no patient receiving tipifarnib doses up to 400 mg BID and bortezomib at 1 g/m2 develop grade 3 toxicities.
Place in Therapy, Conclusions
The enormous progress in molecular biology has revolutionized modern antitumor treatment by unraveling the pathogenetic mechanisms of cancer at the molecular level and thus providing specific targets for pharmacological inhibition.
Farnesyltransferase is such a target for tipifarnib, which is currently the most studied FTI in patients with hematologic malignancies. Patients with AML and late-stage MDS unfit for intensive chemotherapy due to either advanced age or poor performance status constitute the primary target group of tipifarnib. However, despite its favorable toxicity profile and convenient oral route of administration, which allows treatment in the outpatient setting, monotherapy with tipifarnib has shown only modest antileukemic activity. Similarly, the first studies in AML combining tipifarnib with other chemotherapeutic agents did not demonstrate an improvement in overall survival, though only randomized phase III trials will be able to provide definitive answers. Furthermore, the efficacy of the concomitant administration of tipifarnib with the hypomethylating agents azacytidine and decitabine, the only available drugs showing a clear survival advantage in high-risk MDS and AML/MDS patients has not yet been studied. Such a study would be of particular interest as azacitidine and decitabine are considered as differentiating agents, while tipifarnib exerts predominantly antiproliferative effects. Currently, two phase III studies in AML and high-risk MDS patients are evaluating the efficacy of tipifarnib given either as maintenance monotherapy (ECOG, E2902) or induction treatment along with intensive chemotherapy (UK, NCT00454480). Tipifarnib may also offer clinical benefit in other hematologic malignancies, such as CML, MM and NHL, but the available data are too premature to draw conclusions.
One major concern about tipifarnib is that the precise mechanism of action is virtually unknown and clinical responses usually do not correlate with the degree of farnesyltransferase inhibition or with Ras mutational status. As a corollary of this fact, predicting the response to tipifarnib and identifying patients who will benefit most from tipifarnib treatment is not yet possible. Two interesting studies in AML patients sought to identify molecular predictors of response in tipifarnib. In the first study, gene expression profiling from 58 marrow samples from relapsed/refractory AML patients, revealed eight genes that could differentiate responders from non-responders. 118 The expression of the lymphoid blast crisis oncogene (AKAP13) was consistently low in responders and high in non-responders, thus providing the better response prediction with an overall accuracy of 63%. A few months later, the same author reported that the RASGRP1/APTX gene expression ratio predicts with high accuracy the response to tipifarnib both in newly diagnosed and in relapsed/refractory AML patients. 105 RASGRP1 encodes a nucleotide exchange factor that specifically activates RAS and APTX is involved in DNA excision repair, but how these genes confer sensitivity or resistance to tipifarnib remains unclear.
In summary, tipifarnib represents a novel anticancer agent capable of targeting a wide range of cell signaling pathways. Based on preclinical data showing antiproliferative, antiangiogenic and proapoptotic activity, tipifarnib is currently an established candidate for the treatment of various hematological tumors. However, the weak antitumor activity demonstrated in clinical trials has tempered the optimism and led to a turn towards exploring more effective schedules of administration and discovering predictors of response. Considering the safety and ease of administration, it appears that combination regimens of tipifarnib with more potent or diversely acting drugs in older patients with serious comorbidities will eventually be its main application. In any case, additional, well designed, prospective randomized studies are required to establish the definite place of tipifarnib in the treatment of hematologic malignancies.
Disclosure
The authors report no conflicts of interest.