
Abstract
The identification of compounds that specifically inhibit or kill cancer cells without affecting cells from healthy tissues is very challenging but very important for reducing the side effects of current cancer therapies. Hence, there is an urgent need for improved assays allowing the selectivity of a given compound to be monitored directly. The authors present an assay system based on the competitive co-cultivation of an excess of cancer cells with a small fraction of noncancer human indicator cells generating a fluorescence signal. In the absence of a specific anticancer compound, the cancer cells outgrow the indicator cells and abolish the fluorescence signal. In contrast, the presence of specific anticancer drugs (such as Tyrphostin-AG1478 or PLX4720) results in the selective growth of the indicator cells, giving rise to a strong fluorescence signal. Furthermore, the authors show that the nonspecific cytotoxic compound sodium azide kills both cancer and noncancer cells, and no fluorescence signal is obtained. Hence, this assay system favors the selection of compounds that specifically target cancer cells and decreases the probability of selecting nonspecific cytotoxic molecules. Z factors of up to 0.85 were obtained, indicating an excellent assay that can be used for high-throughput screening.
Keywords
Introduction
C
Various human cancers are commonly treated with cytotoxic drugs having a low therapeutic index (the ratio in drug dose associated with a beneficial clinical response versus that causing undesirable effects). The majority of these drugs act by killing cells that are dividing rapidly, one of the main properties of cancers. However, many normal cells, such as blood cells and cells lining the mouth, stomach, and intestine, are also rapidly dividing. Thus, treatment with compounds targeting rapidly dividing cells induces severe side effects such as immunosuppression, cardiotoxicity, hepatotoxicity, nephrotoxicity, and ototoxicity.2,3 These side effects cannot be avoided as long as the anticancer drugs are not specific for cancer cells. The difficulty in identifying anticancer agents targeting only cancer cells stems from the fact that a cancer cell is genetically and phenotypically almost identical to its healthy physiological counterpart.
Assays used to identify potential anticancer agents would ideally predict side effects on noncancer cells to avoid late attrition of hits. Assays used for the screening of anticancer compounds can be divided into two main classes: biochemical and cell-based assays. Biochemical assays use isolated and purified targets playing a critical role in cancer, such as matrix metalloproteinases (MMPs) and kinases.4,5 However, this strategy requires well-characterized drug targets and may lead to the selection of compounds that are inactive within a cellular context due to cell permeability and intracellular stability problems. 6 Conventional cell-based assay systems used to identify anticancer compounds can circumvent these problems. In these systems, phenotypic changes, such as proliferation rates or the upregulation of proteins related to apoptosis of cancer cells, are monitored in response to the compounds being tested. Usually, such cell-based assays monitor the effect of compounds on cancer cells,7,8 leading to false positives in the case of generally cytotoxic compounds and not taking into consideration the effect of the candidate molecule on noncancer cells. Therefore, a primary screen for hits is usually followed by a secondary screen, which determines the degree of cytotoxicity toward normal human cells. However, because cancer cells are genetically almost identical to cells from healthy tissues, most of the compounds initially selected fail in the secondary screen. Hence, assays monitoring the response of cancer cells and healthy cells simultaneously (and therefore revealing selectivity directly) are required.
We describe here a competitive co-cultivation assay, coupling a positive fluorescence signal with the specific inhibition of cancer cells. Previously, a similar competition-based assay has been described for the screening of species-specific antibiotics. 9 Our assay system is based on the co-cultivation of an excess of cancer cells with a small fraction of noncancer cells expressing a reporter gene. The presence of noncancerous cells allows for direct monitoring of undesired cytotoxic effects on these bystander cells. Furthermore, the cancer cells are not genetically modified and can be used directly, as the reporter gene is expressed only in the noncancer cells (the indicator cells). A significant additional advantage of this system is that the assay does not require any detailed knowledge of the cancer type employed or the potential drug targets. Furthermore, the assay system enables the coupling of the specific killing of cancer cells with a positive fluorescent signal.
Materials and Methods
Cells
Indicator cells expressing a membrane-bound and HA-tagged form of tissue plasminogen activator (HEK293T-tPA) were obtained by retroviral transduction of HEK293T cells with MLV (VSV-G) pseudotype particles packaging the encoding vector as described previously. 10
HEK293T-tPA, MCF-7, A375, and WM2664 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, Carlsbad, CA). The media were supplemented with 10% fetal bovine serum (FBS; GIBCO) and 1% penicillin (10 U/µL) streptomycin (104 µg/mL; GIBCO). Cells were incubated at 37 °C under a 5% CO2 atmosphere saturated with water.
Growth curves
In total, 1500 cells of MCF-7 or HEK293T-tPA cells were seeded in 96-well plates in the presence of 100 µL DMEM media supplemented with 10 µM Tyrphostin-AG1478, 100 µM Tyrphostin-AG1478, or 100 µM sodium azide. Similarly, 1500 cells of A375 or WM2664 cells were cultivated in the presence of 1 µM, 5 µM, 10 µM, and 20 µM PLX4720. Three days after exposure to these compounds, the supernatant from all samples was removed, cells were washed twice with a phosphate-buffered saline (PBS) solution (to remove any remaining dead cells) before 150 µL of fresh DMEM media was added. For the next 10 days, cells were counted using a Neubauer counting chamber at an interval of 48 h (in triplicates) by first washing the samples with PBS and subsequently detaching the cells using 0.25% trypsin (GIBCO-BRL, Gaithersburg, MD). Control samples were grown in DMEM without prior exposure to a compound.
Competition-based assays
The assay using puromycin was performed by co-cultivating puromycin-resistant HEK293T-tPA with HEK293T cells in a ratio of 1:10 (HEK293T-tPA:HEK293T). The assay using Tyrphostin-AG1478 (Sigma-Aldrich, St. Louis, MO) was performed by co-cultivating HEK293T-tPA with breast cancer MCF-7 cells in a ratio of 1:10 (HEK293T-tPA:MCF-7). The assay using PLX4720 was performed by co-cultivating HEK 293T-tPA with melanoma cells (A375 or WM2664) in a ratio of 1:10. Cells were detached from cultivation flasks using 0.25% trypsin, centrifuged at 1300 rpm for 6 min, and washed twice with DMEM media before seeding the cells in 96-well plates at a total density of 40 000 cells per well for the HEK293T-tPA/HEK293T co-cultivations (i.e., 40 000 HEK293T cells and 4000 HEK293T-tPA cells) and 60 000 cells per well for the HEK293T-tPA/MCF-7 co-cultivations and the HEK293T-tPA/melanoma cell line co-cultivations in a total volume of 100 µL DMEM. The cell density was determined using a Neubauer counting chamber. Twenty-four hours after seeding the cells, the supernatant was removed, and DMEM media with or without 5 µg/mL puromycin were added. Puromycin containing media was used for the first 3 days before being replaced by fresh DMEM following a washing step (using PBS) to remove dead cells. For the Tyrphostin-AG1478-based assay and the PLX4720-based assay, Tyrphostin-AG1478 was added at a final concentration ranging from 10 to 150 µM, whereas PLX4720 was added at final concentration ranging from 1 to 20 µM. Samples were left for 3 days before being replaced by DMEM following a PBS washing step. After further incubation of the Tyrphostin-AG 1478 samples for 10 days and the PLX4720 samples for 5 days (to allow surviving cells to expand), the supernatant was removed and cells were washed twice with PBS before adding the fluorogenic plasmin substrate HDVLK-Amc (Bachem, Bubendorf, Switzerland) and plasminogen (Roche, Basel, Switzerland) to a final concentration of 1 mM and 1.67 µM, respectively. Fluorescence was measured using excitation and emission wavelengths of 370 nm and 450 nm, respectively, using a Spectramax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
Determination of Z factors
Z factors were calculated using the equation mentioned in Zhang and Chung, 11 where samples with drugs where considered the positive samples and the samples without drugs were considered the negative samples.
Results
Assay validation
The aim of this work was to establish an assay system based on competitive co-cultivation of two different cell types (cancer cells and noncancer indicator cells), in a way that solely the selective death of cancer cells results in a positive fluorescence signal. For this purpose, we chose indicator cells derived from human embryonic kidney cells as noncancer cells (HEK293T-tPA). 10 These cells constitutively express a membrane-bound form of tissue plasminogen activator, allowing the conversion of the nonfluorescent substrate HDVLK-Amc into the fluorescent product aminocoumarin ( Fig. 1 ).
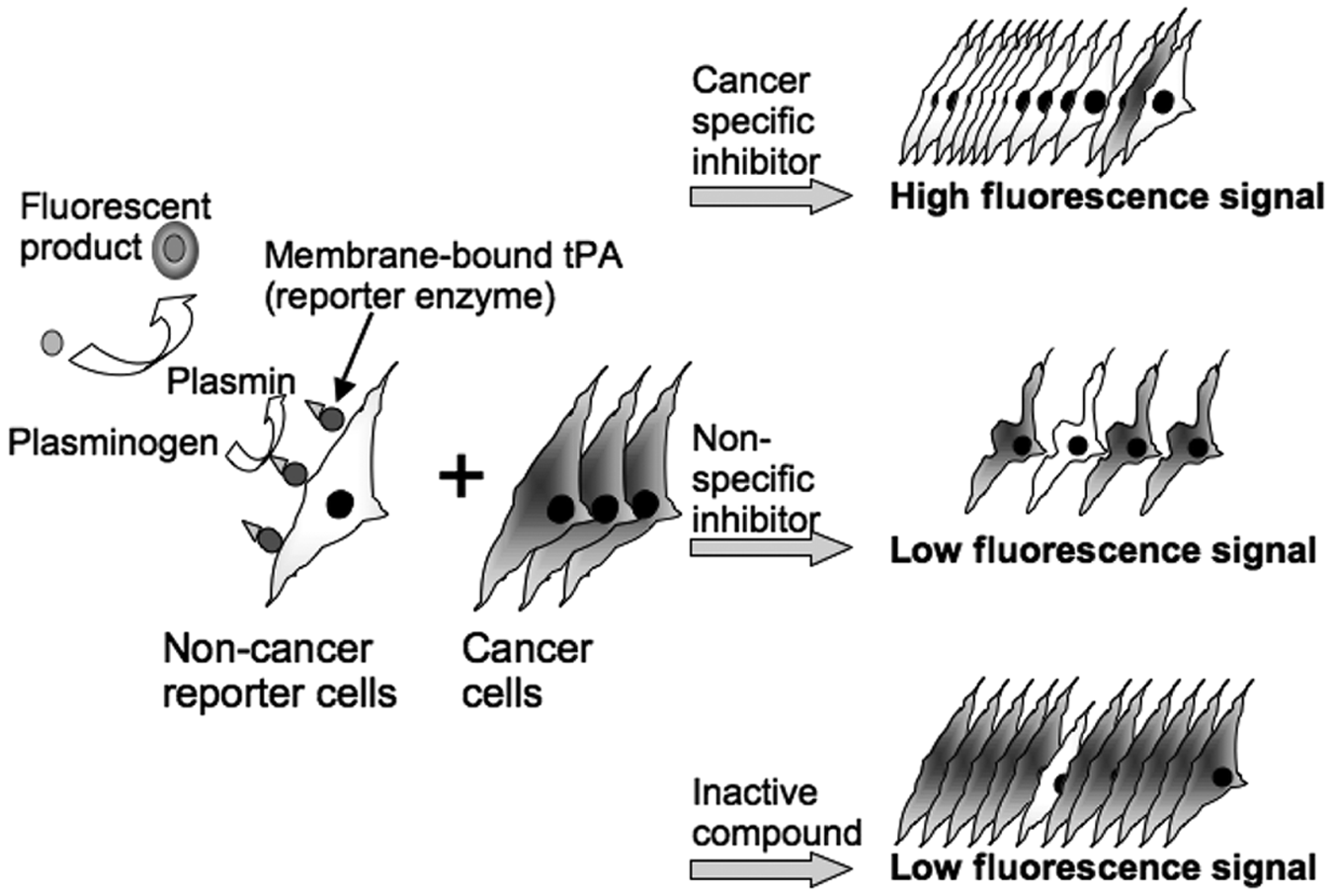
Coupling a positive fluorescence signal to the specific inhibition of cancer cells. The system is based on human noncancer indicator cells displaying a membrane-bound form of tissue plasminogen activator (tPA). This enzyme converts plasminogen into plasmin, which in turn converts a nonfluorescent substrate (HDVLK-Amc) into a fluorescent product. To assay compounds for anticancer properties, a small fraction of indicator cells is competitively co-cultivated with an excess of cancer cells (1:10 ratio). In the absence of an effective anticancer compound, the cancer cells outgrow the indicator cells, thus resulting in a low fluorescence signal. A similar fluorescence readout is obtained for a nonspecific cytotoxic compound where the cancer and the indicator cells are both killed. In contrast, in the presence of a specific anticancer compound, the cancer cells are specifically inhibited and outgrown by the indicator cells, thus resulting in a high fluorescence signal.
In a first step, we performed a proof-of-principle experiment demonstrating that the selective killing of one cell type within a co-culture can indeed be coupled to a positive fluorescence signal. For that purpose, we co-cultivated HEK293T cells (sensitive to the antibiotic puromycin) with the HEK293T-tPA indicator cell line (resistant to puromycin) in the presence of 5 µg/mL puromycin. In regard to the final application of the assay, the HEK293T cells hence served as a model for the cancer cells of interest and puromycin as a drug specifically killing these cells. The co-culture was set up with a starting ratio of 1:10 (HEK293T-tPA:HEK293T) and incubated in the presence and absence of the antibiotic for 3 days. After a further 10 days of co-cultivation (to allow surviving cells to expand), the fluorescence readout was initiated by the addition of plasminogen and the fluorogenic plasmin substrate HDVLK-Amc. Samples without puromycin showed a low fluorescence signal (
Tyrphostin-AG1478 as a model for a cancer-specific compound
Next, to analyze the suitability of the assay system for the selection of truly specific anticancer compounds, the co-cultivation assay was repeated using the already marketed anticancer drug Tyrphostin-AG1478 and MCF-7 cells (a breast cancer cell line) together with the HEK293T-tPA indicator cells.
Tyrphostin-AG1478, which belongs to a family of well-known tyrosine kinase inhibitors used in cancer therapy,
12
was described for the first time in 1988.
13
For cells overexpressing the endothelial growth factor receptor (EGFR), such as the breast cancer cell line MCF-7, Tyrphostin-AG1478 has been reported to inhibit growth and induce apoptosis at concentrations as low as 40 µM.14,15 To further prove the specific effects of this model drug, we first carried out growth studies of homogeneous cultures (MCF-7 and HEK293T-tPA cells separately cultivated) in the presence and absence of 100 µM Tyrphostin-AG1478 (
Dose–response experiments
To demonstrate that the assay allows quantitative dose–response measurements of a given compound, co-cultures of MCF-7 and HEK293T-tPA were exposed to increasing concentrations of Tyrphostin-AG1478 ranging from 10 to 150 µM (10, 25, 50, 75, 100, 125, 150 µM). Increasing concentrations of Tyrphostin-AG1478 correlated with increasing fluorescence signals ( Fig. 2a ). However, for concentrations above 100 µM, the signal intensity decreased, most likely due to cytotoxic effects on HEK293T-tPA cells at high concentrations. Hence, the strongest fluorescence signal was not obtained at maximum (lethal) concentrations but rather at concentrations exhibiting an optimal balance between specific inhibition of cancer cells and cytotoxicity. The results clearly show that the assay system allows monitoring inhibitory effects on cancer cells and the reference cell line simultaneously.
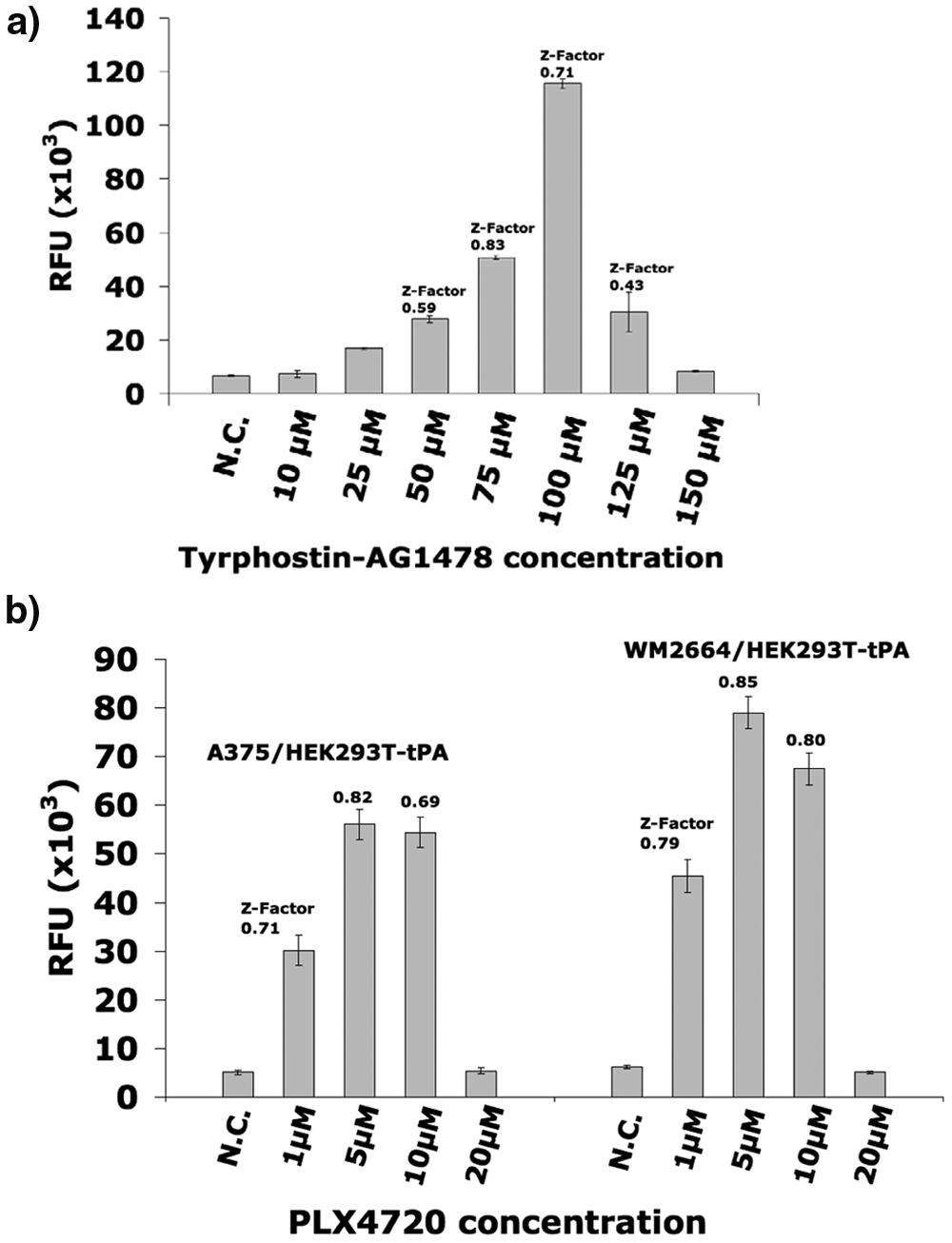
Fluorescence signals obtained from co-cultures (1:10 ratio between indicator and cancer cells) of (
Further assay characterization based on PLX4720, a BRAF-mutant selective cancer-specific compound
To further validate our assay, we decided to co-cultivate melanoma cell lines with our HEK293T-tPA indicator cell line in the presence of PLX4720. PLX4720
16
was first described in 2007 as a potent and selective inhibitor of melanoma cell lines carrying the BRAFV600E mutation, which is the most frequent oncogenic protein kinase mutation known to date. Hence, we have chosen two melanoma cell lines, which are highly sensitive to PLX4720: A375, carrying a BRAFV600E mutation (PLX4720 proliferation GI50 = 0.5 µM), and WM2664, containing the BRAFV600D mutation (PLX4720 proliferation GI50 = 1.5 µM). Growth curves of those melanoma cell lines showed a strong decrease of the growth rates after addition of PLX4720. Samples containing 5 µM PLX4720 grew 100-fold fewer cells compared to samples grown in the absence of the drug (
As a next step, A375 or WM2664 was co-cultivated with HEK293T-tPA cells in the 1:10 ratio of cells used before, in the presence of increasing concentrations of PLX4720 (from 1–20 µM). After a 3-day exposure to the drug, the supernatant was removed and replaced by DMEM for all samples. After a further incubation of 5 days, fluorescence was determined by adding plasminogen and the plasmin substrate HDVLK-Amc. As in the Tyrphostin-AG1478-based assay, increasing concentration of PLX4720 correlated with increasing fluorescence intensity for the PLX4720-sensitive cell lines A375 and WM2664. The highest fluorescence intensity corresponded to samples containing 5 µM PLX4720 ( Fig. 2b ), for which the fluorescence was approximately 11-fold higher than the fluorescence of the untreated samples. For concentrations above 10 µM (20 µM), a drastic decrease in the fluorescence intensity was noticed with both co-cultures, indicating that PLX4720 shows a toxic effect at high concentrations also in BRAF wild-type HEK293T-tPA cells ( Fig. 2b ). The calculations of the Z factor for the concentrations of PLX4720 that exhibited an increase in the fluorescence intensity ranged between 0.71 and 0.85 for the PLX4720-sensitive cells (i.e., A375 and WM2664). Taken together, these results clearly demonstrate that the novel assay system reliably detects specificity of a given drug and clearly allows distinguishing between cells carrying a BRAF mutation and BRAF wild-type cells.
Sodium azide as a model for a nonspecific cytotoxic compound
To demonstrate that this assay system excludes unspecific toxic compounds (inhibiting both cancer and noncancer cells) from the selection, we performed co-cultivation experiments in the presence of different concentrations of sodium azide (0.1 µM to 10 mM), a nonselective highly cytotoxic compound ( Fig. 3 ). The fluorescence of all samples containing sodium azide was as low as the negative control sample. Unlike Tyrphostin-AG1478 or PLX4720, increasing concentrations of sodium azide did not increase the fluorescence intensity when compared to untreated negative control samples. This indicates that sodium azide was killing the breast cancer cells and the melanomas but also abolished the generation of a fluorescence signal by killing the HEK293T-tPA indicator cells, resulting in a low fluorescence signal. The negative control samples also showed a low fluorescence signal because the cancer cells overgrew the HEK293T-tPA indicator cells. Hence, this demonstrates that the novel assay system enables simultaneous monitoring of inhibitory effects on cancer cells as well as cytotoxic effects on the reference cell line. This screening approach thus eliminates false-positive signals from cytotoxic compounds not selective for cancer cells.
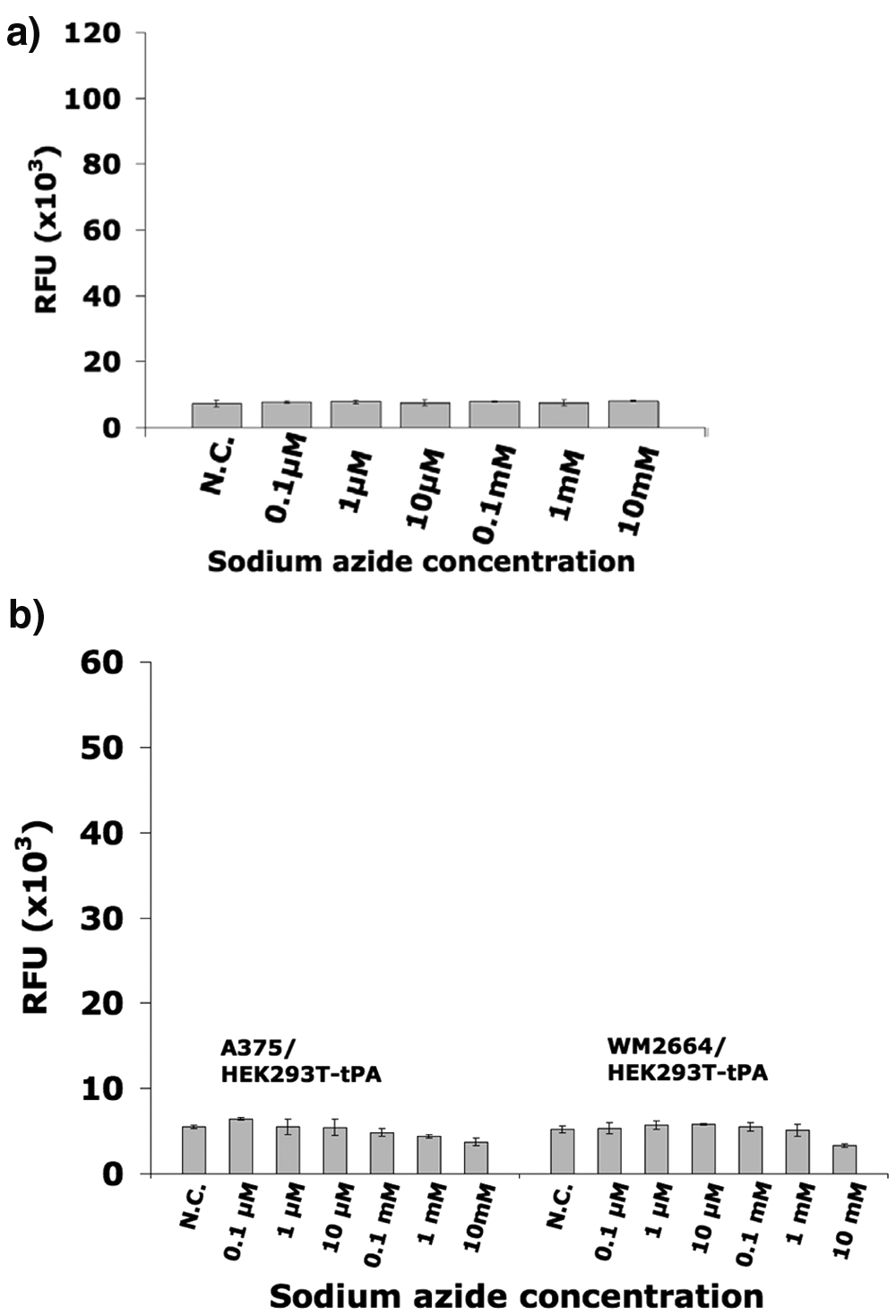
Fluorescence signals obtained from (
To further rule out the possibility that HEK293T-tPA indicator cells simply outgrow the applied cancer cell lines, we assayed co-cultures in the absence of any drug and followed fluorescence development over time (as a measure for the growth rate of the indicator cells under competitive conditions). These experiments show that after a slight increase in fluorescence during the first days, the intensity decreases again to the starting level (
Reliability of the assay
To evaluate the reproducibility of the assay, we performed two completely independent sets of experiments (Run1 and Run2) on 2 different days of both the Tyrphostin-AG1478-based assay and the PLX4720-based assay. Each experiment contained triplicates of all samples for which fluorescence was determined (using Tyrphostin-AG1478 concentrations of 10 to 150 µM, 24 samples per run, or PLX4720 concentrations of 1 to 20 µM, 15 samples per run and cell line). The values obtained for Run1 were plotted against the corresponding values of Run2 ( Fig. 4 ), and a linear correlation was obtained from the data points, indicating a high degree of reproducibility for the assay. The fitted trend line has a coefficient of determination (R2) of 0.95 for the Tyrphostin-AG1478-based assay and 0.91 for the PLX4720-based assays. In parallel, we determined the (mean) relative standard deviation (RSD) between the replicates in one run (intracomparison) and between the two runs (intercomparison). The values obtained were 2.6% (intracomparison) and 6.5% (intercomparison) for the Tyrphostin-AG1478-based assay and 4.5% (intracomparison) and 7.2% (intercomparison) for the PLX4720-based assays, thus demonstrating high reliability and robustness of the assay.
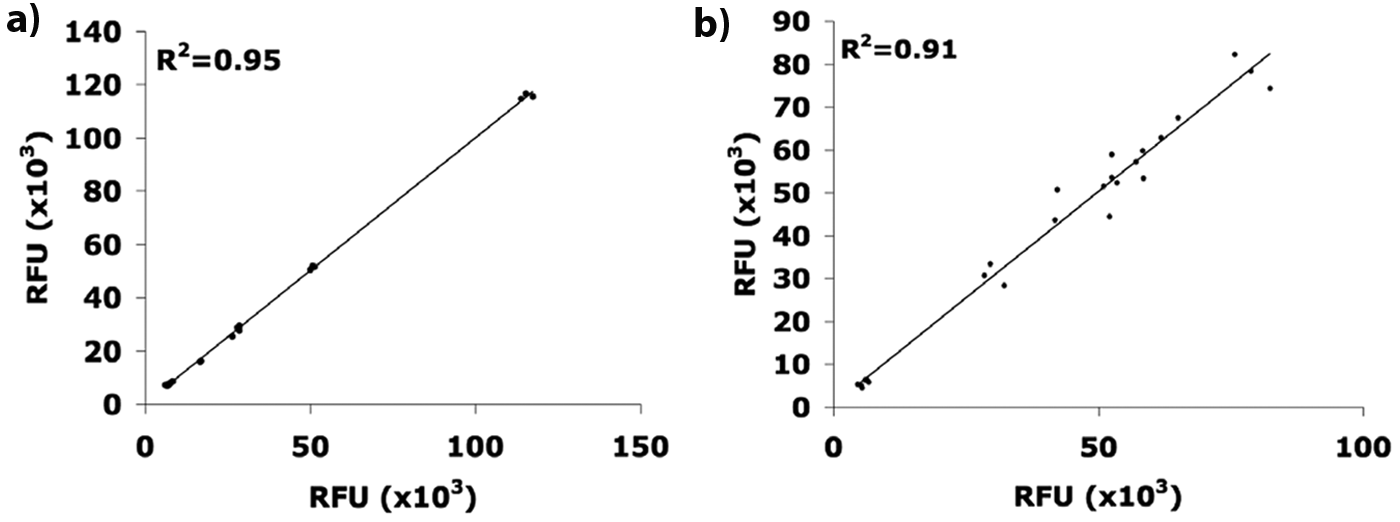
Reproducibility of the co-cultivation assay of (
Discussion
In this proof-of-concept study, we have developed a generic fluorescence assay that couples a positive fluorescence signal with the specific inhibition of cancer cells. The system is based on the competitive co-cultivation of noncancer human indicator cells with the cancer cells of interest. Using Tyrphostin-AG1478 as a model of a cancer-specific compound, we obtained Z factors between 0.59 and 0.83 and an RSD of 2.6% for all samples within one run and 6.5% for equivalent samples between two runs, showing the suitability of this assay system for HTS campaigns. An approximately half-maximal fluorescence intensity (indicating a 50% lethal dose, LD50) was obtained for a Tyrphostin-AG1478 concentration of 75 µM, which is in good agreement with literature: It is reported that 40 µM Tyrphostin-AG1478 induced apoptosis in around 45% of MCF-7 cells. 14 The reliability of the assay was also demonstrated using BRAF mutant melanoma cells and PLX4720, a BRAF mutation-specific drug. In co-cultivation experiments, the BRAF mutant cell lines were strongly outgrown by the BRAF wild-type HEK293T-tPA indicator cell line. Moreover, the high Z factors obtained in these experiments (0.59–0.85) prove that this assay can be used in high-throughput screens to identify compounds selectively killing cancer cells showing mutations of key pathways. Interestingly, a half-maximal fluorescence intensity was obtained at a concentration of approximately 1 µM PLX4720, which is in very good agreement with previous studies reporting GI50 values between 0.5 and 1.5 µM PLX4720 for WM2664 and A375 cells. 16 Taken together, the assay system based on competitive co-cultivation is reliable and offers further advantages: In contrast to conventional assays, adverse side effects of screened compounds and/or cytotoxic concentrations are taken into account directly. We have shown that sodium azide, a toxic compound harming both cancer and noncancer human cells, does not yield a positive fluorescence signal. In line with this, cytotoxic concentrations of Tyrphostin-AG1478 (above 100 µM) and of PLX4720 (above 10 µM) resulted in decreasing fluorescence signals, thus clearly indicating significant damage to noncancer cells. Hence, the novel assay system eliminates the need for an additional cytotoxicity screen subsequent to the primary screen and should thus be of high interest for high-throughput protocols.
A further advantage of this assay system is the fact that it does not require any genetic modifications of the cancer cells of interest because the readout signal is generated by a noncancer indicator cell line (in our model system HEK293T-tPA cells). Hence, there is no need for detailed knowledge about a specific drug target within the cancer cells.
Another interesting feature of the assay is its flexibility in terms of the cancer type against which a compound can be selected, as in theory any cancer cell line can be co-cultured with any type of noncancer indicator cells (e.g., HEK293T-tPA). This feature could also be exploited for personalized therapy by testing cancer cell samples from individuals and optimizing the type and dose of drugs that induce an effective and specific eradication of this cancer before starting the treatment. 17
Footnotes
Acknowledgements
This work was supported by the Ministère de l’Enseignement Supérieur et de la Recherche Français, the Centre National de la Recherche Scientifique (CNRS), and the National Genome Research Network (contract no. 01GS0864) of the German Federal Ministry of Education and Research (BMBF). C.A.M. gratefully acknowledges a Liebig Fellowship of the Fonds der Chemischen Industrie (FCI).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.