
Abstract
The HCV p7 protein is not involved in viral RNA replication but is essential for production of infectious virus. Based on its putative ion channel activity, p7 belongs to a family of viral proteins known as viroporins that oligomerize after insertion into a lipid membrane. To screen for compounds capable of interfering with p7 channel function, a low-throughput liposome-based fluorescent dye permeability assay was modified and converted to a robust high-throughput screening assay. Escherichia coli expressing recombinant p7 were grown in high-density fed-batch fermentation followed by a detergent-free purification using a combination of affinity and reversed-phase chromatography. The phospholipid composition of the liposomes was optimized for both p7 recognition and long-term stability. A counterscreen was developed using the melittin channel-forming peptide to eliminate nonspecific screening hits. The p7 liposome-based assay displayed robust statistics (Z′ > 0.75), and sensitivity to inhibition was confirmed using known inhibitors.
Introduction
I
A less well-studied viral protein is HCV p7. This protein is not involved in HCV RNA replication but has been shown to be essential for viral assembly and release. 1 Viral genomes encoding point mutations that abrogate p7 activity are not infectious in chimpanzees, and such mutations significantly impair in vitro virus production from Huh-7 liver cells. 2,3 Based on its putative ion channel activity, p7 belongs to a family of viral proteins known as viroporins that oligomerize after insertion into a lipid membrane. 4 The p7 monomer consists of 63 amino acids, most of which are hydrophobic, and has endoplasmic reticulum (ER) retention signals. The detergent-solubilized p7 protein has been shown to assemble in a hexameric and heptameric flower-like shape channel structure. 5–7 Each monomer is composed of an N-terminal α-helix followed by a turn and the first transmembrane helix, which is followed by a long cytosolic loop bearing a dibasic motif that connects to a second transmembrane helix. 5
Sensitivity of p7 ion channel activity to inhibition has been demonstrated in vitro using hexamethylene amiloride, adamantanes such as amantadine and rimantadine, and long alkyl chain iminosugars. However, these inhibitors are active against only certain HCV genotypes, and different groups have reported differing sensitivities. 8–11 Ideally, inhibitors would have similar potency against all genotypes. More potent p7 inhibitors are required to assess the potential to inhibit p7 from different HCV genotypes. To develop a robust high-throughput screening (HTS) assay to identify novel p7 inhibitors with improved profiles, we modified a previously reported liposome-based fluorescent dye permeability assay. 12 In this format, a signal is generated after release of fluorescein through p7 pores in the reconstituted liposome. Prior to p7 addition, the fluorescein is self-quenched because of its high concentration within the liposomes.
The assay was converted to an ultra-HTS (uHTS) compatible format by modifying the p7 purification procedure to avoid the use of detergents, which can cause nonspecific leakage from liposomes. The phospholipid composition of the liposomes was optimized for p7 incorporation and long-term stability. Finally, improved p7 handling protocols were developed for optimal activity, and several other parameters were optimized to obtain robust uHTS statistics. The optimized assay was validated by demonstrating sensitivity of the assay signal both to inhibition using known inhibitors and to replacement of wild-type p7 with an inactive, dibasic motif loop variant.
Materials and Methods
Chemicals and reagents
1-(1-Adamantyl)ethylamine hydrochloride (rimantadine), 5(6)-carboxyfluorescein (CF), melittin, pepstatin A, phenylmethylsulfonyl fluoride (PMSF), and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were from Sigma-Aldrich (St. Louis, MO). Antipain and leupeptin were from Bachem (Bubendorf, Switzerland). 2,2,2-Trifluoroethanol (TFE) was from Alfa Aesar (Ward Hill, MA). Cholesterol (Chol), sphingomyelin (SM), L-α-phosphatidic acid (PA), L-α-phosphatidylethanolamine (PE), L-α-phosphatidylserine (PS), L-α-phosphatidylcholine (PC), L-α-phosphatidylinositol (PI), L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) were from Avanti Polar Lipids (Alabaster, AL). N-(n-Nonyl)deoxynojirimycin (NN-DNJ) was from Toronto Research Chemicals (North York, Ontario, Canada). All other chemicals were of analytical grade.
p7 expression system
The codon optimized gene for expression in Escherichia coli encoding the following amino acid sequence, HHHHHHDDDDKALENLVVLNAASVAGAHGILSFLVFFCAAWYIKGRLVPGAAYALYGVWPLLLLLLALPPRAYA, was synthesized by DNA 2.0 (Menlo Park, CA). This sequence comprises an N-terminal His-tag followed by the enterokinase cleavage site and the p7 sequence from HCV genotype 1b Con 1. A SacII site was inserted 5′ of the p7 sequence, and a HindIII site was inserted 3′ of two stop codons. The synthetic gene was subcloned in SacII/HindIII-restricted pET26-Ub to obtain plasmid pETUb-His-EK-p7 for expression in E. coli BL21 (DE3) containing plasmid pCG1 (encoding ubiquitin-specific protease). 13 The K44A/R46A double variant was generated by site-directed mutagenesis.
Production, purification, and characterization of p7
The p7 protein production was performed with a BioFlo 5000 fermenter (New Brunswick Scientific, Edison, NJ) using a high cell–density fed-batch process. 14 The 25-L starting culture consisted of HCDM1 media containing 10 g/L glycerol, 25 µg/mL kanamycin, and 20 µg/mL chloramphenicol. The fermenter was inoculated with BL21(DE3)pCG1/pETUb-His-EK-p7, and cells were grown at 37°C until depletion of the initial glycerol. At that point, HCDF1 feed was initiated and the rate adjusted to maintain growth. When OD600 reached 50, the temperature was reduced to 22°C, and 400 µM isopropyl-β-D-thiogalactopyranoside (IPTG) was added. The culture was incubated for 9 h at 22°C, reaching an end point OD600 of 56. Throughout the procedure, pH was maintained at 7.0 using 15% NH4OH and dissolved oxygen at 20% saturation by sparging with an O2/air mixture. The cells were harvested and frozen at –80°C. This procedure generated a wet cell pellet of 4.2 kg, which was processed batchwise in 1-kg fractions.
One kg of frozen cell pellet was resuspended in 4 L of lysis buffer (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 1 mM TCEP, 10% glycerol, 10 mM imidazole, 5 mg/L pepstatin A, 5 mg/L leupeptin, 5 mg/L antipain, and 1 mM PMSF) at room temperature, using a stir bar, until it appeared to be thawed. All subsequent procedures were performed at 4°C. The solution was homogenized using a motorized marine blade impeller and cycled 3 times in a microfluidizer (M-110Y; Microfluidics, Newton, MA) at 15 000 psi. The lysed extract was centrifuged at 16 000 g for 55 min. The supernatant was batch-adsorbed to 250 mL of Chelating Sepharose FastFlow resin (GE Healthcare, Piscataway, NJ) charged with Ni2+ and equilibrated in IMAC buffer A (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 1 mM TCEP, 10% glycerol, 10 mM imidazole) for 30 min. The resin was then packed into a XK 50/20 column (GE Healthcare) with bottom aspiration using a peristaltic pump at 40 mL/min. The packed column was washed with 4 column volumes (CV) of IMAC buffer A, and a 0%–100% gradient with IMAC buffer B (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 1 mM TCEP, 10% glycerol, 500 mM imidazole) was performed over 8 CV at 10 mL/min using an AKTA Explorer system (GE Healthcare). The protein eluted at 55%–100% IMAC buffer B, and fractions of 14 mL were collected. The fractions (882 mL) were pooled, and 48-mL aliquots were prepared and frozen at –80°C.
Four aliquots (192 mL) were thawed and filtered (0.2 µm), and trifluoroacetic acid (TFA) was added to a final concentration of 0.065%. The solution was then loaded on a Resource RPC 3 column (GE Healthcare), which had been equilibrated in mobile phase A (0.065% TFA in water) at 2 mL/min in 3 batches separated with a 20-mL wash with mobile phase A to remove salts. A 0%–100% gradient with mobile phase B (80% acetonitrile; 0.05% TFA) was performed over 15 CV at 2 mL/min. The protein eluted at 65%–95% mobile phase B, and fractions of 1 mL were collected. The fractions were dried using a SpeedVac system, resuspended in 90% TFE, and stored at –20°C.
Liposome preparation and characterization
Optimized liposomes were composed of 50% PA/20% PE/20% PS/10% PC and encapsulated a solution of 50 mM CF. They were prepared according to the following protocol: 10 mg/mL phospholipids stock solutions in chloroform were mixed in the proper ratio (600 µL PA, 240 µL PE, 240 µL PS, and 120 µL PC) and divided into six 2-mL glass vials, each containing 200 µL of the above mixture. Solutions were dried under a gentle flow of nitrogen and then under vacuum for 4 h. To each vial, 1 mL of 50 mM CF in 10 mM HEPES (pH 7.4) and 100 mM NaCl (HBS) was added, overlaid with argon, and sealed with a Teflon-lined cap. The mixture was shaken for 16–18 h at room temperature (IKA VXR basic Vibrax, 1200 rpm). The resulting liposomes were then extruded 10 times using a 10-mL Lipex Extruder (Northern Lipids, Burnaby, BC, Canada) with a 200-nm filter. Liposomes were washed with 4 × 1 mL HBS by repeated centrifugation (125 000 g, 10°C, 20 min) and resuspension of the pelleted liposomes. At the last step, the pelleted liposomes from each tube were resuspended in 0.5 mL of HBS and pooled to yield 3 mL of liposome suspension. Liposomes were stored at 4°C overlaid with argon. During liposome and assay optimization, other lipids and ratios were tested using the above protocol for liposome preparation. Liposome integrity was evaluated by comparing the fluorescent signals obtained under different conditions versus the signal obtained in the presence of 0.5% Triton X-100, which disrupts the liposomes to give maximal release and dilution of CF, defined as 100% signal. Vesicle size was determined by dynamic light scattering using a Dynapro plate reader model DP-PR-03 (Wyatt Technology, Santa Barbara, CA).
Automated assay
A volume of 5 µL of test compound in HBST (HBS with 1 mM TCEP) plus 1.2% DMSO was delivered using a Matrix 2 × 2 or a Microlab STARlet 384 Hamilton Liquid Handler to Greiner Bio-One (Monroe, NC) FLUOTRAC 200 LoBase Medium Binding Black µClear plates. A volume of 0.5 µL of 8 µM p7 solution in 50% TFE (20×) was added using a MultiDrop Combi nL for which all components were demonstrated to be unaffected by 50% TFE. Positive and negative control wells contained no inhibitors, and negative control wells did not contain p7. Plates were incubated for 10 min at 21–22°C. A volume of 4.5 µL of liposome suspension (~200 µM lipid content in HBST) was added to each well using a MultiDrop Combi. The plates were incubated at 21–22°C for 10 min prior to bottom fluorescence intensity reading using an EnVision (PerkinElmer, Waltham, MA) equipped with a FITC 485 excitation filter, a FITC 535 emission filter, and a 50/50 beam splitter as bottom mirror. Alternatively, during assay optimization, the increase in signal over time was monitored using a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA).
Results and Discussion
Liposome permeability assay principle
In the liposome-based fluorescent dye permeability assay ( Fig. 1A ), a five-fold increase in fluorescence is generated after the addition of p7 to liposomes containing a self-quenching high-concentration CF. A background of ~20% of the maximal fluorescence was observed and could be used to monitor liposome swelling. Liposome integrity was evaluated by comparing the fluorescence signals obtained under different conditions versus the signal obtained in the presence of 0.5% Triton X-100, which disrupts the liposomes to give maximal release and dilution of CF, defined as 100% signal. This maximal signal corresponds to ~2 µM CF. The nonfunctional KR/AA variant was used to demonstrate that the fluorescence release was dependent on a biologically active p7 protein rather than another component of the assay (e.g., detergent).
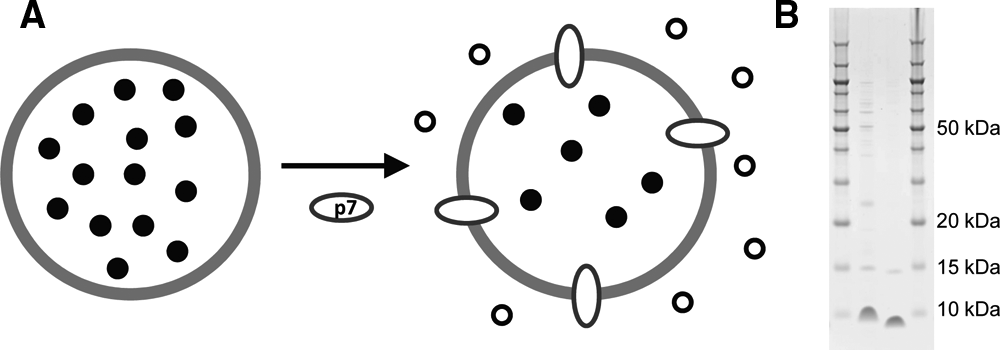
p7 assay format and protein purification. (
Protein purification and stability optimization
In contrast to previously published reports, we found that CF leakage activity from liposomes reconstituted with detergent-purified p7 could not be inhibited by known inhibitors. Moreover, CF leakage was also observed from liposomes formed using the detergent-purified nonfunctional KR/AA variant. These results suggested that variable amounts of residual detergent may be carried over from the purification and contribute to observed activity. This difference from the published results may be due to minor variations between protein sequences or subtle differences between purification protocols. As described in Materials and Methods, a detergent-free procedure was developed to prevent any detergent carryover. Using this procedure, p7 was expressed at high levels (7 mg/OD600/L), but most of the protein was insoluble, and purified protein yields were very low. Yields of purified p7 were 0.025 mg/OD/L and 0.26 mg/OD/L for wt and the KR/AA double variant, respectively. The final product was ≥70% pure as judged by gel electrophoresis ( Fig. 1B ).
After protein purification, p7 was dried, solubilized in the appropriate solvent, and then diluted to generate the stock solution for activity determination. It was observed that p7 solubilized and diluted in methanol was not active when added to liposome-containing mixtures. When dissolved in TFE and diluted into aqueous solution at room temperature prior to liposome addition, p7 rapidly formed aggregates as determined by gel filtration and loss of porin activity. For example, when diluted in HBST prior to liposome addition, the signal dropped by 5% and 30% after incubations of 5 and 15 min, respectively, when compared to p7 diluted in 50% TFE. A minimal content of 50% TFE was required in the p7 stock dilution; if diluted in only 25% TFE, the activity level dropped 47% after 2 h, 60% after 16 h, and 99% after 3 days. In comparison, only a 6% decrease in activity was observed after 3 days of dilution in 50% TFE. Notably, a recent study demonstrated that the maximal α-helical content of p7 is reached at 50% TFE. 5 Thus, maintaining p7 in a highly α-helical conformation in the stock dilution may be important to maintain activity.
Optimization of liposome composition
For leakage assays, liposomes were prepared by encapsulating a solution of 50 mM CF (a self-quenching concentration) in 10 mM HEPES-OH (pH 7.4), 100 mM NaCl. As described in Table 1 , a high level of PA in liposomes was required for p7 porin activity. This was in good agreement with a previous report in which p7-derived peptides were shown to bind to negatively charged lipids. 15 It appeared that the optimal liposome size was 200 nm. It was also observed that 100-nm liposomes had similar stability during storage but had lower activity relative to 200-nm liposomes. In initial experiments, liposomes formed with 50% PA were unstable. However, stable liposomes with optimal activity were obtained by adding lipids to the mixture that mimic natural membrane contents (PE, PS, PC, and SM). Several lipid combinations were evaluated, and a composition of 50% PA, 20% PE, 20% PS, and 10% PC was found to be optimal with respect to liposome formation, storage stability, and p7 incorporation. Therefore, this lipid composition was selected for further experiments. Furthermore, when diluted to the assay concentration, such liposomes were stable at room temperature for at least 40 h.
Stability of Liposome Stock Solutions and p7 Leakage Activity of Different Liposome Compositions
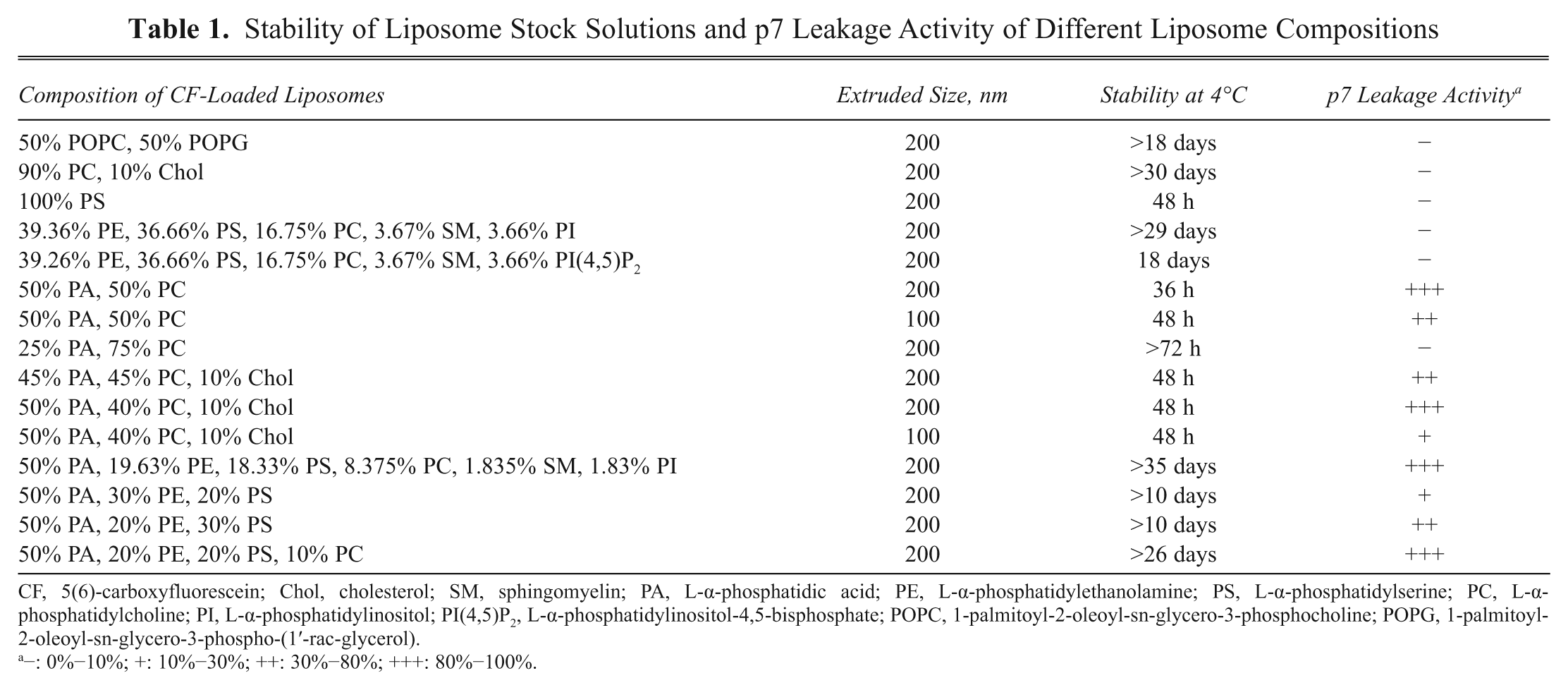
CF, 5(6)-carboxyfluorescein; Chol, cholesterol; SM, sphingomyelin; PA, L-α-phosphatidic acid; PE, L-α-phosphatidylethanolamine; PS, L-α-phosphatidylserine; PC, L-α-phosphatidylcholine; PI, L-α-phosphatidylinositol; PI(4,5)P2, L-α-phosphatidylinositol-4,5-bisphosphate; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPG, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol).
−: 0%−10%; +: 10%−30%; ++: 30%−80%; +++: 80%−100%.
Optimization of p7 activity
As described above, in the final assay format, the stock solution of p7 was maintained at 50% TFE for optimal activity. However, when mixing p7 with liposomes, we found that it was important to dilute TFE as much as possible (>10-fold dilution). In preliminary experiments using unoptimized liposome size and content, we found using dynamic light scattering that increasing amounts of TFE (in the absence of p7) caused an increase in liposome radius from 88 nm in HBST to 101 nm in 2.5% TFE and 108 nm in 5% TFE ( Fig. 2A ). This increase in liposome size led to an increase in background fluorescence signal from the liposomes by slightly reducing the self-quenching of the CF signal. A similar but less significant effect was also observed with increasing DMSO concentration ( Fig. 2A ). Final concentrations of 0.6% DMSO and 2.5% TFE were selected to provide the best balance of liposome stability and assay signal/background ratio. The required 1:20 dilution of the p7 stock solution was obtained by delivering 0.5 µL of p7 into 9.5 µL of assay buffer containing test compound, using a Multidrop Combi nL. Using these conditions, preincubation of p7 with compounds for 5 to 20 min prior to addition of liposomes led to a slight 10%–20% decrease in activity versus no preincubation but had no effect on inhibition parameters. Because we felt that a p7-test compound preincubation would be desirable to maximize the potential of inhibitor identification, a 10-min p7-test compound preincubation time prior to liposome addition was chosen.
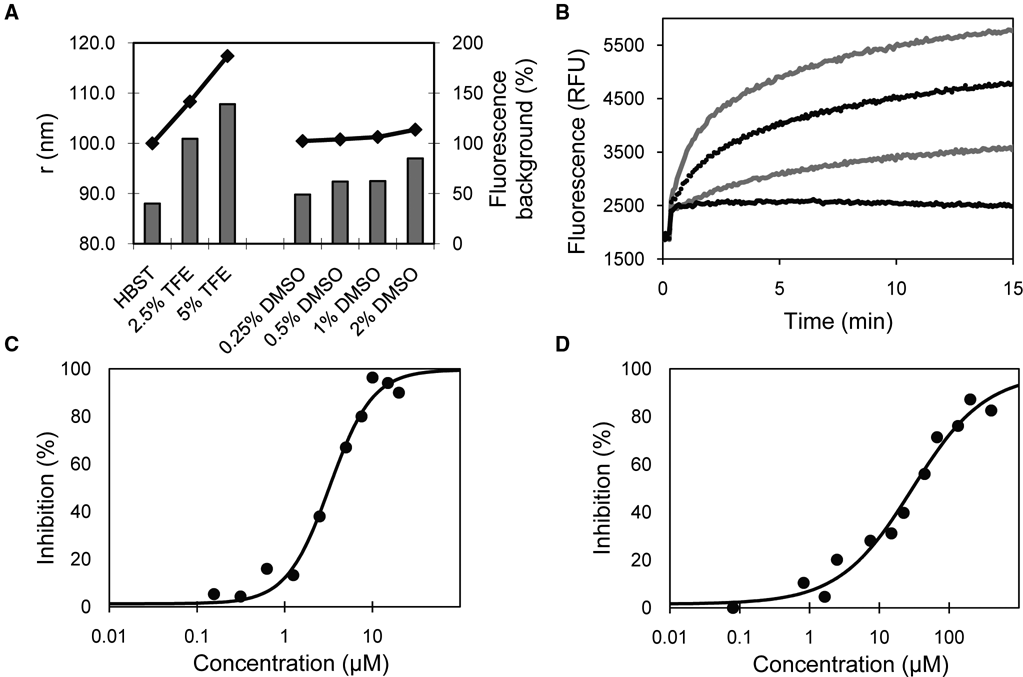
Optimization and validation of the p7 liposome leakage assay. (
Mixing of p7 to liposomes resulted in a time-dependent increase in signal to a plateau obtained after ~20 min. The maximal signal obtained in the presence of 0.5% Triton X-100 was ~11 000 relative fluorescent units (RFU) after 15 min. Increasing concentrations of p7 resulted in a higher initial rate and a higher plateau in the fluorescence signal ( Fig. 2B ). An increase in plateau with increasing p7 concentration suggests that p7 was not incorporated in all liposomes. A final concentration of 0.4 µM p7 and an incubation time of 10 min with liposomes were chosen for end-point reads to be used for screening. In comparison, addition of 1 µM of the KR/AA double variant resulted in only a small immediate 15% increase in signal above background with no further increase in signal. This is identical to the addition of buffer/TFE alone and is probably due to swelling of the liposomes from the TFE addition (lower black line trace; Fig. 2B ). This initial immediate increase in fluorescence due to liposome swelling was also observed in all the p7 traces and can be subtracted when evaluating the kinetics of pore formation. Using the initial rate of fluorescence increase or end-point read after 10 min led to the same inhibition parameters. When reading on the EnVision, consistently higher signal to background (S/B) and Z′ were obtained with bottom-read of clear-bottom plates relative to top-read of opaque plates. The use of nonbinding surface plates destabilized liposomes and caused CF release in the absence of p7, most likely due to the coating agents used. Under optimized assay conditions, IC50 values of 3.3 ± 0.4 µM and 30 ± 3 µM were obtained for reported inhibitors NN-DNJ and rimantadine, respectively ( Figs. 2C , 2D ).
Use of the assay for compound screening
A subset of 3520 compounds (ten 384-well plates) randomly selected from the full corporate screening collection was tested at 3 µg/mL using the automated assay format. An average S/B of 2.4 ± 0.1 and Z′ of 0.76 ± 0.03 for each plate were obtained. When using a cutoff of 42% of control (POC) (3SD), a 1.8% hit rate was observed for a total of 62 hits ( Fig. 3 ). False positives due to compound interference are common when using nM concentrations of CF fluorescence in HTS. However, given the very high fluorescence signal observed in this assay format, interference was not a concern. Only 0.06% of test compounds reached 1% of the total fluorescence signal.
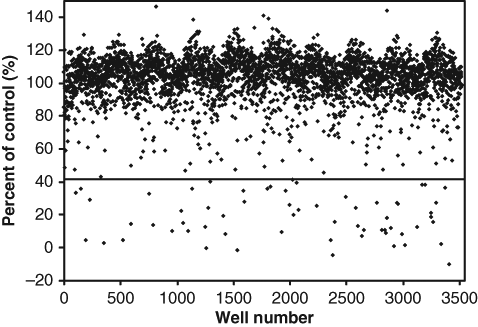
Screening of a subset of the full corporate compound collection. In total, 3520 compounds were tested at 3 µg/mL. When using 3 SD as a hit cutoff, a value of 42% of control (POC; dotted line) was obtained, and a 1.8% hit rate was observed.
Melittin counterscreen
A counterscreen to eliminate nonspecific inhibitors was developed with melittin, a 26-residue structurally dissimilar, strongly basic channel-forming peptide that is the principal component of honey bee venom. This counterscreen allows the differentiation of potentially specific versus nonspecific p7 inhibitors and also serves to rule out any other assay-related artifacts. The counterscreen was tested against the same 3520-compound set under the same conditions as the p7 assay except that a concentration of 1.4 µM melittin was used to obtain an equivalent signal. An average S/B of 4.8 ± 0.2 and a Z′ of 0.8 ± 0.1 were obtained. Forty-three hits were identified using a cutoff of 64 POC (3 SD); only 5 of these hits were also present in the p7 hit list. This result demonstrates the weak sensitivity of this assay to fluorescence artifacts and also suggests that the p7 assay has a high potential to indentify specific p7 inhibitors.
Conclusion
A p7 liposome permeability assay was optimized for uHTS inhibitor screening. In this assay format, a signal was generated by the increase in fluorescence after release of the CF dye from the pores formed by p7. Prior to p7 addition, the CF dye was self-quenched because of its high concentration within liposomes. We have shown that the high level of fluorescence generated in this specific assay format decreases the degree of compound interference that is often observed in fluorescence-based assays. Sensitivity to inhibition was confirmed using known inhibitors as well as a subset of the full screening collection. The identification of specific inhibitors of p7 using this uHTS-compatible assay will facilitate the discovery of novel HCV antivirals.
Footnotes
Acknowledgements
The High-Throughput Biology group of Boehringer Ingelheim Pharmaceuticals (Ridgefield, CT) is acknowledged for suggestions during assay optimization and for demonstrating stability of the Multidrop Combi nL to 50% TFE.