
Abstract
Hepatitis C virus (HCV) is a considerable global health problem for which new classes of therapeutics are needed. The authors developed a high-throughput assay to identify compounds that selectively block translation initiation from the HCV internal ribosome entry site (HCV IRES). Rabbit reticulocyte lysate conditions were optimized to faithfully report on authentic HCV IRES-dependent translation relative to a 5′ capped mRNA control. The authors screened a library of ~430,000 small molecules for IRES inhibition, leading to ~1700 initial hits. After secondary counterscreening, the vast majority of hits proved to be luciferase and general translation inhibitors. Despite well-optimized in vitro translation conditions, in the end, the authors found no selective HCV IRES inhibitors but did discover a new scaffold of general translation inhibitor. The analysis of these molecules, as well we the finding that a large fraction of false positives resulted from off-target effects, highlights the challenges inherent in screens for RNA-specific inhibitors.
Introduction
H
The 5′-untranslated region (5′-UTR) of the HCV genome, a positive-sense, single-stranded RNA, is attractive as an antiviral target due to its importance in the viral life cycle. In particular, nucleotides (nts) 40-372 comprise an internal ribosome entry site (IRES) by forming a specific secondary and tertiary RNA structure essential for viral protein synthesis. 4,5 Both the structure and mechanism of the IRES have been studied extensively, revealing some of the molecular events that lead to translation initiation. 6 The IRES RNA binds directly to the human 40S ribosomal subunit and to eukaryotic initiation factor 3 (eIF3). 7,8 The IRES thereby bypasses the need for most initiation factors involved in typical cellular translation initiation, such as the cap-binding eIF4F complex and scanning-associated factors eIFs 1, 1A, and 4A. 7 Specific steps in this pathway can be blocked by mutation or deletion of particular regions of the IRES RNA. 9-11
The IRES is one of the most conserved regions of the HCV genome because it must function both in ribosome recruitment to the 5′ end of the plus-strand RNA and in viral replication from the 3′ end of the minus-strand. 2 Despite its functional importance, the IRES is a challenging drug target as it has no enzymatic activity or explicit active site but instead uses an extended surface to interact with both the 40S ribosomal subunit and eIF3. 12,13 Although inhibition of HCV IRES translation with an antisense oligonucleotide to disrupt the structure of a key region of the IRES has been preliminarily successful, 14,15 direct inhibition of IRES binding to 40S subunits or to eIF3 may be difficult to achieve with a small molecule.
Importantly, however, the IRES is more than a molecular scaffold since mutations that do not affect its affinity for the host translation machinery can nonetheless dramatically reduce translation efficiency. 8,11 For example, conformational flexibility of the domain II hairpin is required to induce a conformational change in the 40S subunit, 13 a necessary precursor to formation of active 80S ribosomes. An attractive possibility is that small molecules can be found that specifically disrupt local structures or conformational dynamics of the RNA required for IRES function.
In an effort to discover such small-molecule inhibitors of the HCV IRES, we developed a high-throughput assay based on IRES-dependent in vitro translation in rabbit reticulocyte lysate. Conditions for this assay were designed to report on authentic HCV IRES translation relative to a 5′ capped mRNA control. We screened a library of 426,736 small molecules for IRES inhibitors, leading to ~1700 initial hits of which 59 appeared promising after secondary assays. That a large number of these molecules proved to be off-target luciferase inhibitors underscores the challenges in screens for compounds that recognize RNA targets and in using enzymes as readouts for activity.
Materials and Methods
Reagents
Restriction endonucleases, calf intestinal alkaline phosphatase, and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA). [35S]methionine (>1000 Ci/mmol) was obtained from GE Life Sciences (Boston, MA). Plasmid DNA preparation, restriction enzyme digestion, agarose gel electrophoresis of DNA and RNA, DNA ligation, and bacterial transformations were carried out using standard methods. 16
Plasmid construction and RNA transcription
A DNA fragment encoding the HCV IRES sequence was generated by PCR using a genotype 1a HCV IRES construct (H77 strain, 17 nts 40-372) as the template. The resulting DNA fragment was ligated into the EcoRI and BamHI restriction sites of pUC19 to form the parent plasmid for all subsequent constructs. Derivative plasmids encoding the IRES with domain II deleted (ΔdomII, nts 40-119 deleted) were generated by using QuikChange mutagenesis (Stratagene, La Jolla, CA). The firefly (FF) and Renilla (RN) luciferase reporter genes were amplified from pGL3 and pRL-TK, respectively (Promega, Madison, WI), and cloned between the BamHI and HindIII restriction sites of pUC19. Capped messages had a template-encoded poly(A) tail of 62 nt. All constructs were verified by DNA sequencing. IRES and 5′ capped RNAs were prepared by in vitro transcription (Megascript and mMESSAGE T7 system, respectively; Ambion, Austin, TX) according to the manufacturer’s instructions. The 5′ capped messages were generated with a GTP:m7GpppGTP ratio of 1:10 to ensure >90% capping efficiency.
In vitro translation
In vitro translations were performed in rabbit reticulocyte lysate, and translation activity was measured using either a luciferase reporter assay or radiolabeling with [35 S]methionine, according to the manufacturers’ instructions (Promega and Life Technologies, Carlsbad, CA). Unless otherwise indicated, in vitro translations were carried out in 15 µL reactions, containing 2 µL of nuclease-treated rabbit reticulocyte lysate (RRL; 0.5 µL Ambion + 1.5 µL Promega; the composition of RRL was optimized for translation efficiency and accuracy; data not shown), 1 ng/µL and 3 ng/µL of IRES-RN, and 5′ cap-FF reporter mRNAs, respectively, and 5 ng/µL polyC RNA as a carrier. Translation reactions were adjusted to final concentrations of 2.6 mM Mg(OAc)2, 45 mM KCl, and 90 mM KOAc and also contained 1 mM amino acids, 1% DMSO, and 0.1% complete protease inhibitor (Roche, Basel, Switzerland). Translation reactions were incubated at 30°C for 90 min and terminated by the addition of puromycin to a final concentration of 20 µM.
For high-throughput screening (HTS), reactions were scaled proportionally to 5 µL total volume, and the positive controls for IRES-RN and 5′ cap-FF translation inhibition were a DNA oligonucleotide complementary to the IIId loop of the IRES (10 µM) and puromycin (20 µM), respectively. The IIId oligo sequence used was 5′-ACCCAACACTACTCGGC-3′. 14 A combination of DualGlo, BrightGlo, and Dual Luciferase Assay System kits (Promega) was used in preliminary assays, as noted in figure legends, to measure firefly and Renilla luciferase activities from the same well. The DualGlo luciferase assay kit (Promega) was selected for HTS due to its long half-life of luminescence and was used according to the manufacturer’s instructions with minor adjustments. Luminescence was measured by TopCount (PerkinElmer, Waltham, MA) for 96-well plates and by EnVision for 384-well plates (PerkinElmer). For the secondary enzyme interference screen, in vitro translation reactions were performed as above but in the absence of compounds. After quenching translation reactions with puromycin, compounds were added and incubated with the translated enzymes for 30 min at room temperature before measuring luciferase activity.
For [35S]methionine incorporation experiments, translation reactions were performed as above, except that methionine was omitted from the amino acid mixture, and 10 µCi of [35S]methionine was added. Aliquots from translation mixtures were mixed with sodium dodecyl sulfate (SDS) sample buffer, boiled for 2 min, and resolved on a 10% SDS polyacrylamide gel. Gels were fixed in 10% methanol and 7.5% acetic acid and treated with ENHANCE (New England Nuclear, Boston, MA). The level of translation of FF luciferase (61 kDa) and RN luciferase (36 kDa) was quantified by phosphorimaging and densitometry using ImageQuant TL (Molecular Dynamics, Sunnyvale, CA).
Compound library and liquid handling
The chemical library for HTS consisted of 426,736 compounds (Gilead Sciences, Foster City, CA). Liquid handling protocols were optimized for distribution of (a) the compound library (Bravo, Velocity11, Menlo Park, CA), (b) translation mix to minimize formation of air bubbles (Deerac, Tecan, Männedorf, Switzerland), (c) RNA templates to ensure precise dispensing at small volumes (Deerac, Labcyte, Sunnyvale, CA), and (d) luciferase substrates (microFill, BioTek, Winooski, VT).
Data analysis
Data analysis was carried out by GraphPad Prizm5.1 (GraphPad Software, La Jolla, CA) and Spotfire (TIBCO, Somerville, MA). The Z factor was calculated as previously described. 18 The normalized percentage of inhibition values (NPI, %) was calculated for each HTS plate by setting the average signal of the negative control (1% DMSO) as 0% inhibition and the average signal of the positive control (IIId oligo or puromycin) as 100% inhibition.
Results
Translation inhibition screen design
A robust functional assay was established to screen for small-molecule inhibitors that block HCV IRES-mediated translation. To enable independent assessment of 5′ cap-dependent and cap-independent translation initiation from the same well, we designed 2 monocistronic mRNAs encoding distinct luciferase enzymes, one driven by a 5′ cap and the other by the HCV IRES ( Fig. 1A ; genotype 1a, H77 strain, nts 40-372). The IRES primary sequence and tertiary structure are highly conserved among all 6 genotypes, 19,20 making it likely that inhibitors found against this genotype would have pan-genotypic activity. For the translation of the reporter constructs, we considered both HeLa cell extract and RRL. Although HeLa translation extracts are reported to faithfully recapitulate the translation behavior observed in cells, 21 low activity prevented obtaining sufficient signal for the statistical analysis required for a high-throughput screen. In contrast, unmodified RRL has limited 5′ cap-dependence and low sensitivity to mutations in the HCV IRES 22,23,this study but produces high levels of translation. These observations led us to focus on optimization of RRL as the system in which to conduct inhibitor screens.
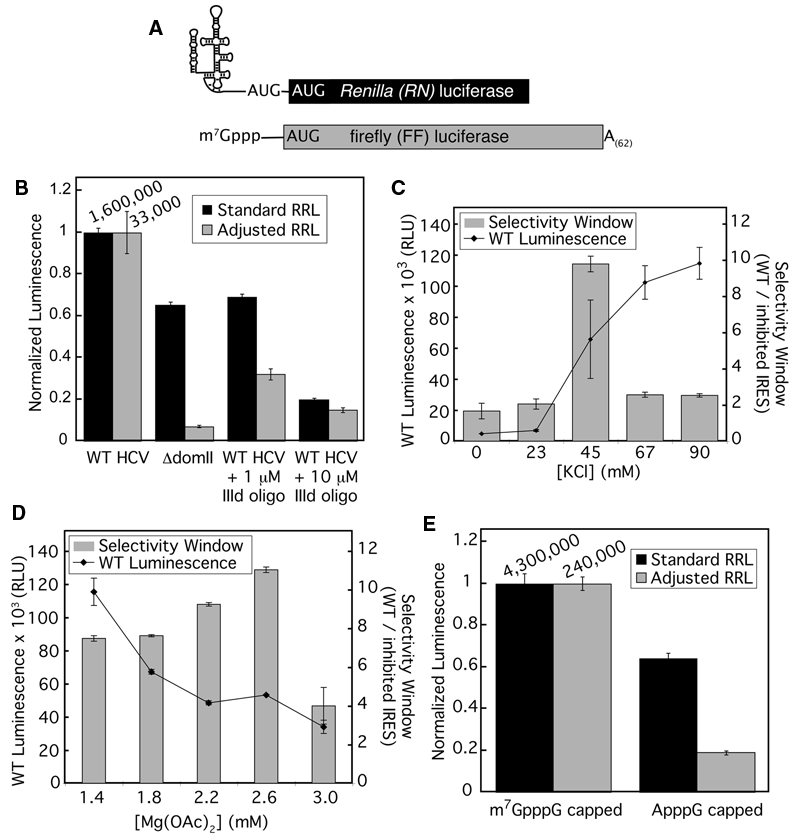
Optimization of rabbit reticulocyte lysate for authentic translation of hepatitis C virus (HCV) internal ribosome entry site (IRES) with a firefly luciferase reporter. (
In light of previous results showing that the salt concentrations in RRL could be adjusted to stimulate faithful 5′ cap-dependent scanning, 24 we examined the effect of RRL salt concentration on the fidelity of HCV IRES-mediated translation. To distinguish IRES-dependent from promiscuous translation, we used 2 established methods for inhibiting HCV IRES function: (a) deletion of domain II (nts 40-119, referred to as ΔdomII 9 ) and (b) inhibition with a DNA oligonucleotide complementary to the IIId loop of the IRES, a region required for recruitment of the 40S ribosomal subunit (referred to as IIId oligo). 14
Under standard RRL conditions (79 mM KOAc, 0.5 mM MgCl2; Promega), a reporter mRNA containing the ΔdomII IRES was translated with 65% of the activity of an mRNA containing the wild-type IRES (WT; Fig. 1B , black columns). By comparison, the ΔdomII mRNA has been shown to have only 3%, 2%, and 20% activity in mice, HeLa cells, and HeLa S10 extracts, respectively. 11,21 Notably, the inhibitory activity of the IIId oligo against IRES-mediated translation was also limited under these conditions ( Fig. 1B ). When the salt concentration of the RRL reactions was adjusted to a previously identified condition (2.2 mM Mg2+, 45 mM KCl, 90 mM KOAc), 24 the ΔdomII-containing mRNA showed a significant reduction in translation activity (7% of the wild-type IRES-containing mRNA). Furthermore, the IIId oligo was more effective at inhibiting IRES-driven translation in the adjusted RRL when compared to standard RRL ( Fig. 1B ). The ratio of luciferase activities from translation of WT IRES-containing mRNA versus either ΔdomII IRES or WT IRES in the presence of the IIId oligo defined the “selectivity window” of the assay. This selectivity window can be considered to report on both the authenticity of IRES translation and our ability to detect inhibition of this translation. Further optimization of KCl ( Fig. 1C ) and Mg(OAc)2 ( Fig. 1D ) showed that the largest selectivity window of 11-fold occurred at 45 mM KCl and 2.6 mM Mg(OAc)2.
Under these adjusted RRL conditions, a 5′ cap-containing mRNA was translated ~5-fold better than the same mRNA containing a nonphysiological cap ( Fig. 1E ). Thus, the adjusted RRL displayed increased fidelity for both IRES- and 5′ cap-dependent translation. Although the overall activity of the salt-adjusted RRL was decreased relative to unaltered conditions (nearly 20-fold reduction for 5′ capped mRNA and ~50-fold for IRES-containing mRNA), these activity levels were significantly higher than those observed with HeLa extracts (which showed at least 20-fold lower activity in the presence of greater than 100-fold more mRNA; data not shown).
Reporter mRNA choice and assay optimization
To identify IRES-specific inhibitors, we designed a high-throughput screen using 2 reporter mRNAs encoding distinct luciferase enzymes, RN and FF. To maximize the signal-to-noise ratio in the primary screen, we wanted each well to contain as much of the 2 monocistronic mRNAs as possible without introducing competition between them, which could result from one reporter preferentially monopolizing ribosomes and other translation factors in the extract. Titrations of 5′ capped-FF and IRES-RN mRNAs were performed using the optimized salt conditions established above, measuring the signal from one reporter at a time. As IRES-RN mRNA concentration was increased to moderate to high concentrations, the signal from 5′ capped-FF translation dropped (
Fig. 2A
; note the vertical spread), which was indicative of interference between the 2 messages. Similarly, the highest concentrations of 5′ capped-FF mRNA interfered with the signal from IRES-RN translation (
Fig. 2B
), although competition in this direction did not appear to be as strong. Significant interference at high concentrations of either reporter mRNA was also demonstrated for the converse reporter pair (IRES-FF and 5′ capped-RN; see
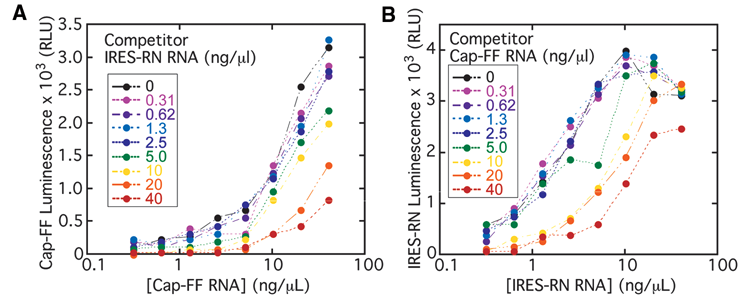
Two-dimensional titrations of internal ribosome entry site–Renilla (IRES-RN) and 5′ cap-firefly (FF) mRNAs in adjusted rabbit reticulocyte lysate (RRL). The concentration of each of the 2 mRNAs was varied between 0 and 40 ng/µL, and the absolute luminescence activities of (
The RRL-based translation assay conditions were further adjusted to be suitable for HTS of potential small-molecule inhibitors. Because the small molecules to be tested were dissolved in DMSO, we examined the effect of this organic solvent on the activity and selectivity window of the translation reactions. Although the signal from IRES-FF mRNA steadily decreased as DMSO concentration increased ( Fig. 3A ), the selectivity window between WT IRES- and ΔdomII IRES-containing mRNAs, representing the faithfulness of the extract, remained greater than 6.5 in the presence of up to 2% DMSO. These data showed that the final DMSO concentration of 1% was compatible with this assay.
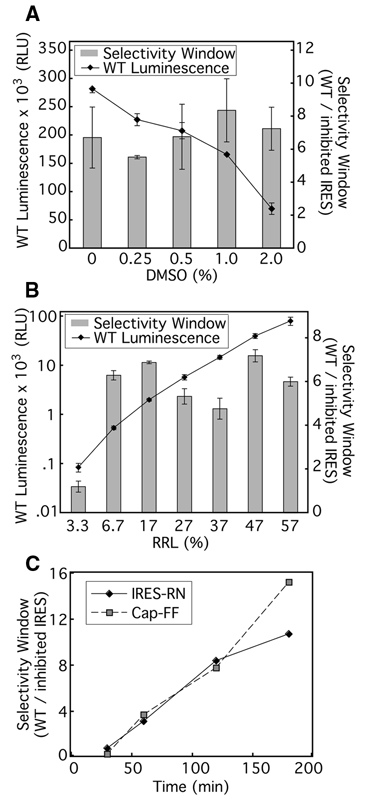
Optimization of rabbit reticulocyte lysate (RRL) translation assay for high-throughput screening (HTS). The absolute luminescence signal of wild-type (WT) internal ribosome entry site–firefly (IRES-FF) mRNA is shown in relative light units (RLU). (
It was also possible to significantly reduce the amount of RRL used in the assay for implementation in the high-throughput screen. The selectivity window and the signal from WT IRES-containing mRNA were measured as RRL concentration was varied from 3.3% to 57% (vol/vol), and the overall salt concentration was kept constant. The luminescence signal decreased by ~1000-fold over the range of titration, but the selectivity window remained relatively stable from ~57% down to 6.7% ( Fig. 3B ). Lastly, it was important to ensure the linearity of reaction kinetics. At 13% RRL, the condition chosen for HTS, the 5′ capped-FF signal remained linear for at least 3 h, whereas the IRES-RN signal began to plateau at ~120 min ( Fig. 3C ). Thus, an incubation time of 90 min was chosen for the high-throughput screen as it gave substantial signal and was within the linear range. In the final format for the primary screen, 5 µL translation reactions containing 13% RRL (Ambion:Promega = 1:3) were incubated at 30°C for 90 min.
Primary screen
Using the assay conditions outlined above, a total of 426,736 compounds (average molecular weight [MW] ~550) were tested in a primary screen. The Z factor was determined for each 384-well plate. The average Z factors for 5′ cap-FF and IRES-RN reporters were 0.71 and 0.62, respectively (ranging between 0.5 and 0.86; see
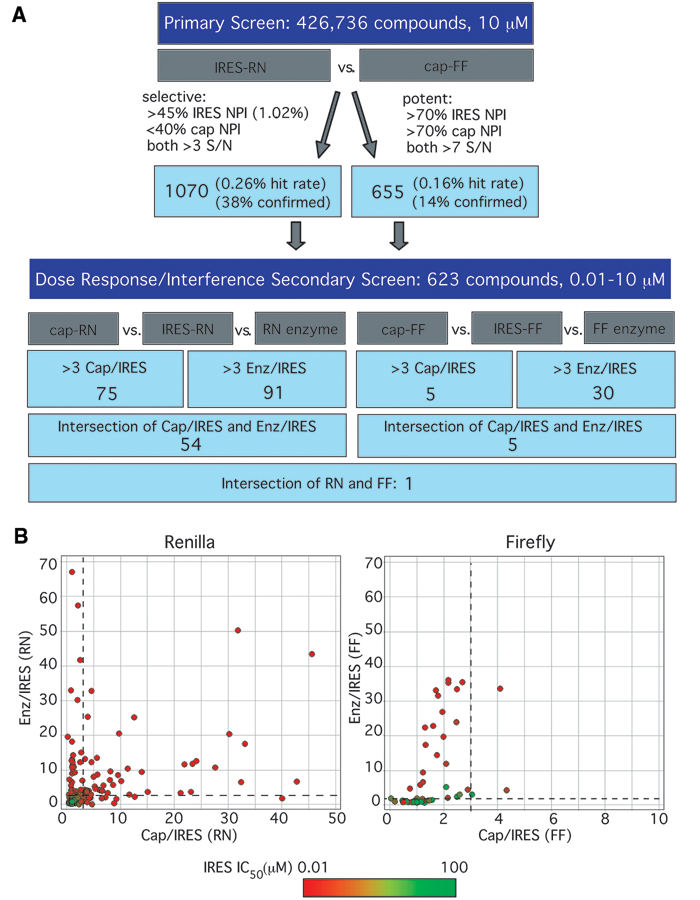
Results of high-throughput screen. (
Secondary screen
Of these ~1700 primary hit compounds, a representative group of 623 compounds was purchased for follow-up dose-response assays. These compounds were chosen based on commercial availability and their drug-like nature, determined by an automated in silico triage based on molecular weight (<600 Daltons), lack of reactive groups (e.g., methylating groups), and solubility (calculated partition coefficient, log P <5). The goals of these assays were (a) to define the selectivity window between IRES- and 5′ cap-dependent inhibition, even for potent hits, and (b) to distinguish authentic IRES inhibitors from compounds that inhibit Renilla luciferase preferentially over firefly luciferase. Two formats of secondary screens were used. One was essentially identical to the primary screen except it consisted of the opposite pair of reporters: IRES-FF and 5′ capped-RN mRNA. The second test was a luciferase enzyme interference assay in which the translation of IRES-RN and 5′ capped-FF was performed as in the primary screen but in the absence of any compounds. Once the translation reaction was quenched with puromycin, compounds were added and incubated with the translated enzymes before measuring luciferase activity. In total, these assays yielded 6 sets of data: IC50 values for each compound against IRES-RN, 5′ capped-RN, IRES-FF, 5′ capped-FF (compounds present during translation reactions), and IC50 values against Renilla enzyme and firefly enzyme (compounds added after the translation of enzyme). Data for these dose-response follow-up assays were plotted as the selectivity index (SI; ratio of IC50) calculated for the 5′ cap/IRES and the enzyme (Enz)/IRES comparisons made for each luciferase ( Fig. 4B ).
The vast majority of compounds in both the Renilla and the firefly assays showed a selectivity index of less than 3 for both Enz/IRES and 5′ cap/IRES, falling into the lower left-hand quadrant in Figure 4B . Some of these compounds, in green, had very high IC50s against the IRES-containing message, suggesting that they were simply false positives from the primary screen. Many other compounds, in red or orange, had low IC50s against the IRES-containing message, indicating that they were correctly identified by the primary screen as inhibitors of the IRES-containing message. These compounds are therefore most likely luciferase inhibitors that equally inhibit the luciferase signal, whether it is driven by the IRES or 5′ cap or comes directly from the enzyme.
There were also a large number of compounds present in the upper left-hand quadrants of the plots in Figure 4B , showing selectivity for IRES-driven translation over the enzyme alone but not over 5′ cap-dependent translation. Many of these molecules are likely to be general translation inhibitors capable of blocking translation driven either by the 5′ cap or the IRES. Consistent with this inhibition acting at the level of general translation, these compounds behaved similarly against both RN and FF reporters: of the 21 compounds that were found in this upper left-hand quadrant for FF and the 34 compounds in this quadrant for RN, 18 overlap. It is also possible that some of these compounds are general luciferase inhibitors that were not hits in the enzyme-alone assays due to unavoidable differences in the setup of this assay (see Discussion).
We chose compounds that had a selectivity index greater than 3 for both Enz/IRES and 5′ cap/IRES as potential IRES-selective inhibitors. These compounds fell into the upper right quadrant of either plot in Figure 4B . As the screening cascade shows ( Fig. 4A ), many more compounds met this criterion with the Renilla reporter than the firefly reporter. Of these compounds, only one appeared to be a selective inhibitor with both reporter enzymes (GS-036984, Fig. 5A ).
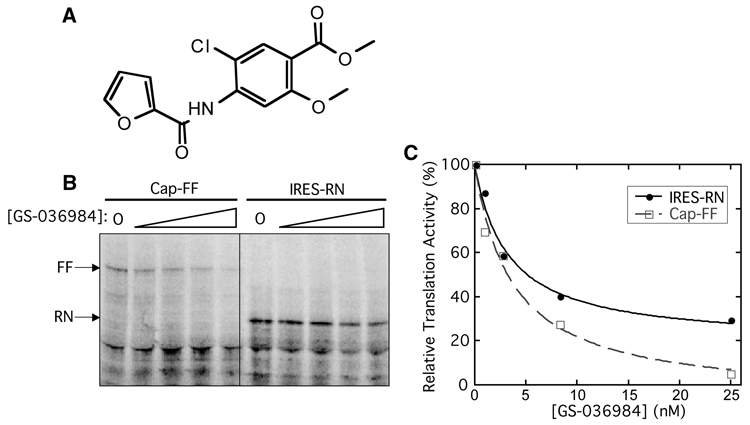
Nonselective inhibition of GS-036984 against internal ribosome entry site (IRES)– and 5′ cap-dependent translation. (
Validation assay
Although GS-036984 appeared to be an IRES-selective inhibitor after the secondary screens using both FF and RN reporters, the selectivity index of 5′ cap/IRES (RN) for this compound was 3.9, barely meeting the cutoff of 3. Therefore, we also wanted to validate this compound’s activity in an assay completely independent of luciferase activity. [35S]methionine incorporation followed by SDS–polyacrylamide gel electrophoresis (PAGE) analysis was used to observe translation of the IRES-RN and 5′ capped-FF messages in the presence of GS-036984 ( Fig. 5B ). Contrary to expectations from the screen, this compound was found to inhibit translation from both 5′ cap-FF mRNA and IRES-RN reporters and thus is in fact a general translation inhibitor ( Fig. 5C ; see Discussion).
We also examined the activity of the 59 compounds that were identified as selective IRES inhibitors with either FF or RN reporters, but not both, using the [35S]methionine incorporation translation assay. One additional compound related to the scaffold above also inhibited both 5′ cap- and IRES-driven translation. To the best of our knowledge, these compounds represent a new scaffold of general translation inhibitor. The other compounds tested, however, failed to inhibit translation of either message (data not shown).
Discussion
The HCV IRES RNA is an attractive drug target due to its central role in regulating viral protein synthesis and pan-genotypic sequence conservation. In this study, we screened a small-molecule library for specific inhibitors of HCV IRES function. The results of this work highlight both successes in high-throughput assay development and challenges inherent in identifying compounds that recognize RNA targets.
Previous work established that direct IRES binding by an antisense oligonucleotide can block IRES-mediated protein synthesis. 14 Although this nucleic acid–based candidate (ISIS 14803) reached phase I clinical trials, limited on-target activity and aminotransferase flares led to a halt of clinical development. 15 In addition, there have been other attempts to identify IRES-specific inhibitors by high-throughput screens with smaller libraries. Fragment-based screening using mass spectrometry has identified small-molecule binders to domain II of the IRES that are inhibitors of an HCV replicon assay. 25 A dual luciferase-based HTS assay in Krebs extracts with a bicistronic reporter did not reveal any IRES-specific compounds. 26 We sought to identify new chemical scaffolds that directly target IRES function and could be further optimized to yield potent oral antiviral agents.
A screening assay was developed using RRL supplemented with additional salts, which improved the fidelity of both cap-dependent and HCV IRES-mediated translation. These results are analogous to what has previously been seen with the encephalomyocarditis virus (EMCV) IRES (although the EMCV IRES requires a different set of translation initiation factors from the HCV IRES). 27 The use of 2 monocistronic reporters (one IRES driven and one 5′ cap driven) enabled each reporter’s signal to be maximized independently while avoiding competition between the 2 reporter templates, which cannot be achieved using a single bicistronic mRNA. We noted that in general, many more compounds in the secondary screen inhibited the RN enzyme than the FF enzyme, and no compounds inhibited both. This result suggests that the primary screen was effective at excluding general luciferase inhibitors, such as competitive inhibitors of the ATP substrate or protein aggregants. However, ~3% of the primary hits appeared to be general translation inhibitors (in the upper left-hand quadrant of Fig. 4B ), which ideally would have been eliminated in the primary screen.
Due to issues with luciferase interference, an [35S]methionine incorporation validation assay was essential to verify the inhibition activity of the 59 compounds that appeared to be selective IRES inhibitors in the secondary screen with either the firefly or Renilla reporter. Two of these compounds were found to be general translation inhibitors and, to the best of our knowledge, represent a new chemical scaffold of general translation inhibitors. The compound GS-036984 was selected from the primary screen as a potent hit, consistent with it inhibiting both 5′ cap- and IRES-dependent translation. The selectivity indices determined in the secondary screen for 5′ cap/IRES were barely above the cutoff of 3.0 (3.9 with the Renilla reporter and 3.3 with the firefly reporter). Although this low selectivity suggested a potential preference for inhibiting the IRES, the direct visualization of translation products without interference of luciferase reporters clearly showed that this compound inhibits both 5′ cap-dependent and IRES-dependent translation ( Fig. 5B , C ).
In the [35S]methionine incorporation validation assay, 57 of 59 compounds identified in the secondary luciferase screen showed no inhibition against either IRES- or 5′ cap-dependent translation. This observation suggests that these compounds are likely to be direct inhibitors of the Renilla luciferase enzyme. Several scaffolds emerged from the compounds showing selective Renilla luciferase inhibition, such as para-amino-sulfonamides, sulfanyltriazoles, and dihydropyrrolones. The 10 most potent Renilla-specific inhibitors identified in the screen are shown in
The relative abundance of luciferase inhibitors identified in this study reflects the fact that it is much simpler chemically for a compound to block an enzyme active site than to bind to a macromolecular surface and inhibit a conformational change or an intermolecular interaction (note that a large fraction [~40%] of current drug targets are enzymes, and many other drug targets also naturally bind to small molecules). 28 Our secondary counterscreens eliminated most but not all of the facile and prevalent direct inhibitors of Renilla luciferase. The high level of Renilla luciferase inhibitors selected in this screen suggests there is an inherent problem in using an enzymatic reporter assay as a means to screen for compounds that interact with a particularly challenging target, such as a structured RNA, as the reporter enzyme is itself a classical target of small molecules.
To expand our ability to devise new and diverse drugs, the scope of small-molecule activities must be expanded to nontraditional targets, such as influencing macromolecular interactions and conformations, and efforts are under way to expand the diversity of small molecules in screening libraries. 29 Based on our experience with enzyme inhibition masking the desired RNA-based inhibition, assays for new functions would do well to avoid traditional small-molecule targets as readouts for activity. Fluorescence, as opposed to luminescence, may be an alternative approach to couple translation activity to a spectroscopic signal. For example, members of the green fluorescence protein family do not have a defined active site for binding small molecules and may therefore be less subject to direct reporter inhibition. A dual-reporter screen could be conducted with a pair of red and green fluorescent proteins, although spectroscopic interference from heterocyclic compounds in the library would need to be considered. 30 In addition, in an effort to reduce enzyme interference, the active site of firefly luciferase has been redesigned and evolved to discourage small molecules binding to the luciferin and ATP pockets. This new enzyme, “Ultra-Glo,” shows a marked loss of inhibition from several scaffolds that inhibited the original firefly luciferase. 31
Other strategies for identifying RNA-binding small molecules as inhibitors that do not require enzymatic readouts, such as fragment-based or in silico screening, could also be considered as alterative approaches. 25,32,33 A fluorescence resonance energy transfer (FRET)–based screen for small-molecule binders to RNA has previously been used to find compounds that bind to and stabilize the HCV IRES IIId loop, 34 but whether these compounds are effective inhibitors of HCV IRES translation has yet to be determined. As a complement to such small-molecule binding screens, it is certainly attractive to envision direct functional screens to look for inhibitors of IRES-mediated translation. However, the present study shows that such assays are not as straightforward as they may first appear.
In this study, we validated the use of rabbit reticulocyte lysate with increased salt concentrations as a system to study the effects of mutations and inhibitors of the HCV IRES. The current screen has provided insights into how deleterious direct enzyme inhibition can be to an otherwise robust high-throughput assay for a difficult target, such as the HCV IRES. In addition, a new scaffold has been identified of a general eukaryotic translation inhibitor, the mechanism of which will be interesting to investigate in the future.
Footnotes
Acknowledgements
We thank members of the Doudna laboratory for helpful discussions and comments on the manuscript; S. Coyle, A. Law, and B. Reid for assistance with data collection; and Gilead IRES team members, M. Desai, M. McGrath, R. Sakowicz, S. Swaminathan, and J. Ward, for insightful discussion and comments on project implementation. This work was supported by a program project grant from the National Institutes of Health and a research gift generously provided by Gilead, Inc. (to J.A.D.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.