
Abstract
DNA gyrase, a type II topoisomerase that introduces negative supercoils into DNA, is a validated antibacterial drug target. The holoenzyme is composed of 2 subunits, gyrase A (GyrA) and gyrase B (GyrB), which form a functional A2B2 heterotetramer required for bacterial viability. A novel fluorescence polarization (FP) assay has been developed and optimized to detect inhibitors that bind to the adenosine triphosphate (ATP) binding domain of GyrB. Guided by the crystal structure of the natural product novobiocin bound to GyrB, a novel novobiocin–Texas Red probe (Novo-TRX) was designed and synthesized for use in a high-throughput FP assay. The binding kinetics of the interaction of Novo-TRX with GyrB from Francisella tularensis has been characterized, as well as the effect of common buffer additives on the interaction. The assay was developed into a 21-µL, 384-well assay format and has been validated for use in high-throughput screening against a collection of Food and Drug Administration–approved compounds. The assay performed with an average Z′ factor of 0.80 and was able to identify GyrB inhibitors from a screening library.
Introduction
DNA
Because the bacterial gyrase holoenzyme has been the subject of multiple drug discovery efforts, many assays exist to measure its activity. Assays used for general studies of the holoenzyme as well as many high-throughput screens measure the ability of the enzyme to convert relaxed DNA into supercoiled DNA. Most of these studies use an assay that couples ATP hydrolysis to nicotinamide adenine dinucleotide (NADH), resulting in a measureable colorimetric change. 6,9,10 Similar assays directly measure the total level of supercoiled DNA using agarose gel separations or fluorescent dyes. 11-13 Cell-based assays that measure the level of DNA damage have also been used to measure gyrase activity. 14 Although all of these assays can be used to measure the activity of the gyrase holoenzyme, they often require multiple addition steps, cannot separate GyrA from GyrB inhibitors, and do not focus on the ATP binding domain. One assay has been described that measures the direct binding of [3H]dihydronovobiocin to a biotin-labeled 43-kDa fragment of GyrB using a scintillation proximity assay (SPA). 15 Although the SPA directly examines the ATP binding domain, an assay that uses fluorescence rather than radioactivity would be better suited for high-throughput screening (HTS).
Fluorescence polarization (FP) is a homogeneous assay that can be used to measure the binding interaction between molecules. 16 FP is based on the principle that a fluorophore excited by polarized light will also emit polarized light. Molecular motion, which is dependent on the size of the molecule, causes depolarization of the light by radiating at a different direction than the incident light. A small unbound fluorescent probe rotates rapidly and maintains low levels of polarization after excitation. If the fluorescent probe binds to a larger molecule, such as a protein, forming a stable complex, the bound probe rotates more slowly and increases the amount of polarized light. Binding is directly related to the polarization level of the sample: an unbound fluorescent probe has low FP, and a bound fluorescent probe has high FP. The FP assay is well suited for measuring the interaction between molecules in real time and is commonly used in HTS. 17
This article presents the development and optimization of a novel FP assay to detect competitive inhibitors of the ATP binding domain of Francisella tularensis GyrB. We have designed and synthesized a novel fluorescent probe by covalently attaching a Texas Red fluorophore to novobiocin (Novo-TRX) guided by the GyrB/novobiocin crystal structure (Protein Data Bank entry 1KIJ). 18 Experiments were performed to develop the FP assay and optimize the use of Novo-TRX to measure the competition for binding to the ATP binding domain of F. tularensis GyrB. We have determined the kinetics of the interaction of Novo-TRX with GyrB as well as the effect of common buffer additives on the interaction. The assay was also validated for use in HTS for inhibitors of the ATP binding domain by screening a small library of Food and Drug Administration (FDA)–approved compounds. This screen identified a known GyrB inhibitor as well as 4 members of the anthracycline family of cancer therapeutics (doxorubicin, idarubicin, epirubicin, and daunorubicin).
Materials and Methods
Reagents
All chemicals were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO) unless otherwise noted.
Synthesis of Novo-TRX
The Novo-TRX probe was synthesized by attaching a reactive secondary amine to novobiocin through a Mannich reaction followed by conjugation to the Texas Red–X fluorescent dye. To produce the reactive novobiocin amine intermediate, novobiocin sodium (400 mg, 0.62 mmol), paraformaldehyde (10.5 mg, 0.69 mmol), methyl-amine (471 µL, 0.94 mmol from 2M tetrahydrofuran [THF] solution), and acetic acid (56 µL, 0.94 mmol) were sealed in a glass microwave reactor vial with 4 mL of anhydrous THF. After the mixture was heated for 3 h at 115 °C, the solvent was removed under reduced pressure. The crude reaction was purified by preparative high-performance liquid chromatography (HPLC) to yield 115 mg of the pure product, which was confirmed by liquid chromatography/mass spectrometry (LC/MS) and 1H–nuclear magnetic resonance (NMR) analysis. The novobiocin amine intermediate (3.7 mg, 4.6 µmol) was dissolved in dry dimethylformamide (DMF; 200 µL) before Texas Red–X SE (2.5 mg, 3.0 µmol; Invitrogen, Carlsbad, CA) and triethylamine (50 µL) were added. The reaction was shaken for 2 h at room temperature, and then the entire mixture was directly injected onto the preparative HPLC system running with acetonitrile and water (0.1% TFA) to yield the pure Novo-TRX probe. The structure of Novo-TRX was confirmed by LC/MS and 1H-NMR analysis.
Overexpression plasmids
GyrB was cloned into a pET28 expression plasmid (Novagen, Madison, WI) by polymerase chain reaction (PCR) from genomic DNA isolated from F. tularensis Schu S4. Primers were selected to add an amino-terminal His6-tag. GyrB was cloned using the forward primer 5′-ATATAGCTAGCATG TTAATGTCTGAGAATAAAGCTTATGAC and the reverse primer 5′-ATATACTCGAGTTATTAA-ACGTCTAAGTTAA CCACATTAAGAG, which also added NheI and XhoI restriction enzyme sites, respectively. The sequence of GyrB was confirmed to be identical to the published source. 19
Protein expression and purification
F. tularensis GyrB was produced from Escherichia coli BL21 Star (DE3) (Invitrogen) that carried the GyrB pET28 expression vector as described above. Briefly, GyrB was grown in Luria-Bertani (LB) media at 37 °C until an optical density (OD)600 of 0.4 was reached and then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Bacteria were grown for 3 h at 30 °C after induction and then collected through centrifugation. The cell pellet was resuspended in TGE (50 mM Tris-HCl [pH 7.9], 5% glycerol, and 0.5 mM ethylenediaminetetraacetic acid [EDTA]) and incubated with 250 µg/mL lysozyme for 20 min at 4 °C. Bacteria were lysed using sonication (4 × 30 s, 70% amplitude) and then centrifuged for 25 min at 22,000 g to remove cell debris.
GyrB was isolated from the supernatant fraction of the E. coli lysate by a multistep purification procedure using the AKTA Explorer (GE Healthcare, Piscataway, NJ). To begin, the supernatant was loaded onto a 5-mL HiTrap Heparin column (GE Healthcare) equilibrated with TGE. Unbound material was washed from the column, and protein was eluted with a 0-1M NaCl gradient over 10 column volumes (CV) with a flow rate of 4 mL/min, and 4-mL fractions were collected. Fractions containing GyrB were identified using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (see
Equilibrium binding experiment
Association binding experiments were performed to determine the time necessary for the binding of Novo-TRX and GyrB to reach equilibrium. These experiments were performed in quadruplicate in a volume of 20 µL of the assay buffer (25 mM NaCl, 50 mM Tris-HCl [pH 7.9], 0.5 mM EDTA, 5 mM MgCl2, 5 mM DTT, 5% glycerol) in black flat-bottom polystyrene NBS 384-well microplates (Corning, Corning, NY). To initiate the experiment, an equal volume of 2× Novo-TRX was added to 2× GyrB to produce a final concentration of 40 nM Novo-TRX and 25, 50, or 100 nM GyrB. The level of FP was determined using an Analyst HT plate reader (Molecular Devices, Sunnyvale, CA) by exciting at 560 nm with polarized light through a Q595 long-pass dichroic mirror and measuring the amount of parallel and perpendicular light at 645 nm with medium attenuation and an acquisition time of 100 ms. The binding of Novo-TRX to GyrB was measured every 30 s for 70 cycles. Upon completion of the association experiments, a 125-fold excess of unlabeled novobiocin (10 µM final concentration) was added, and the change in polarization was monitored every 30 s for 70 cycles to measure the dissociation. The dissociation data were fit using a 1-phase exponential decay equation, Y = Span * exp(−K * X) + Plateau, by GraphPad Prism 4 (GraphPad Software, San Diego, CA) to determine the average half-life of dissociation.
Saturation binding experiment
Binding experiments using a constant concentration of Novo-TRX and increasing concentrations of GyrB were performed in quadruplicate to optimize the window of the assay and to determine the amount of GyrB needed to saturate the binding of Novo-TRX. A method was developed that closely mimics the desired order of addition used in the high control samples of the HTS to measure binding. A serial dilution of 3× GyrB (0-2500 nM final concentration) was made in the assay buffer and mixed with an equal volume (7 µL) of 3X Novo-TRX (40 nM final concentration). The mixture was then diluted with the assay buffer + 15% DMSO to result in the final concentrations of Novo-TRX, GyrB, and 5% DMSO. The samples were incubated for 1 h, and the FP was measured as described above. To determine the concentration resulting in 50% saturation (EC50), the data were fit using the sigmoidal dose-response equation, Y = Bottom + (Top – Bottom)/(1 + 10^(LogEC50 – X)), in GraphPad Prism 4.
Solvent susceptibility experiment
Standard additives for drug screening were tested for their effect on the binding of 40 nM Novo-TRX to 40 nM GyrB. The following components were tested: DMSO, Triton X-100, Tween-20, and BSA. Quadruplicate serial dilutions of a 2× stock of each component were made (10 µL final volume) in black flat-bottom polystyrene NBS 384-well microplates (Corning). To measure the FP, 15 µL of a 2× complex (80 nM Novo-TRX, 80 nM GyrB) in 2× assay buffer (50 mM NaCl, 100 mM Tris-HCl [pH 7.9], 1.0 mM EDTA, 10 mM MgCl2, 10 mM DTT, 10% glycerol) was added to 15 µL of a 2× serial dilution of the various components (20 µL total volume, final concentration of 40 nM Novo-TRX/40 nM GyrB in assay buffer) and incubated covered for 1 h at 25 °C (room temperature). After the incubation, the FP signal was measured as described above.
HTS of a small chemical library
A small chemical library comprising 1040 FDA-approved drugs (1 replicate of each) and 80 additional antibiotics (3 replicates of each) was used to validate the use of the GyrB FP assay in identifying inhibitors of the ATP binding pocket through HTS. The library was screened twice using different preparations of GyrB. Compounds were screened in a 384-well format at a final concentration of 10 µM. Each plate was composed of 320 test compounds, 32 negative controls (5% DMSO), and 32 positive controls (10 µM novobiocin). To perform the screen, 14 µL of a 1.5× master mix of 60 nM Novo-TRX/60 nM GyrB was added to all the wells using the WellMate bulk liquid dispenser (Thermo Scientific). The PlateMate Plus liquid handler (Thermo Scientific) was then used to add 7 µL of the positive and negative controls to the plate, and then 7 µL of the test compounds in 15% DMSO was added to the plates, resulting in a final concentration of 40 nM Novo-TRX, 40 nM GyrB, 10 µM compound, and 5% DMSO. The plates were incubated for 1 h at room temperature, and the FP signal was measured as described above. The Z′ factor, Z factor, and signal-to-noise (S/N) and signal-to-background (S/B) ratios were calculated as described by Zhang et al. 20
Dose-response experiment
The ability of the hits identified in the HTS to disrupt the interaction of Novo-TRX with GyrB was confirmed using an FP dose-response experiment. The potent small-molecule inhibitor, N-ethyl-N′-[7-(2-pyridinyl)-5-(3-pyridinyl)-1H-benzimidazol-2-yl]-urea (Vertex-13), was synthesized as described 21 and used as an additional control to validate the assay. In brief, 40 nM Novo-TRX was used along with 40 nM GyrB in black 384-well plates. Quadruplicate stocks of the compounds were made by first diluting the compound (stored in 100% DMSO) 10-fold into the assay buffer + 15% DMSO, then serially diluting 3-fold with assay buffer + 15% DMSO to yield 3× compound concentration; 7 µL of the compounds was then mixed with 14 µL of a 1.5× mixture of Novo-TRX/GyrB to give a final volume of 21 µL and 5% DMSO. The FP was read after a 1-h incubation at room temperature as described above. The background signal was subtracted and the FP signal was normalized to the DMSO control. Data were plotted and fitted, and the concentrations resulting in 50% inhibition (IC50) were calculated using the sigmoidal dose-response equation in the GraphPad Prism 4 software.
Gyrase activity assay
Gyrase activity was measured using the purified E. coli DNA Gyrase and Relaxed DNA kit (Topogen, Port Orange, FL) according to the manufacturer’s instructions. Briefly, the E. coli gyrase holoenzyme was incubated with 0.5 µg of the relaxed pHOT1 DNA for 1 h at 37 °C, and the level of supercoiling was measured by agarose gel electrophoresis. DNA was visualized with ethidium bromide staining.
Results and Discussion
Design of FP assay for GyrB
Although enzymatic assays for GyrB exist, an equilibrium binding assay suitable for HTS would have several advantages. Since the holoenzyme is a heterotetramer (A2B2), enzymatic assays require the recombinant production of both the GyrA and GyrB subunits and then subsequent mixing to form the active complex. The enzymatic assays have the potential to identify inhibitors at multiple binding sites. A homogeneous equilibrium binding assay, such as FP, requires minimal reagent addition steps and can be used to efficiently identify inhibitors of the GyrB ATP binding site in an HTS format.
The assay design strategy was to use the natural product novobiocin, which is known to tightly bind into the ATP binding site of GyrB, and tether a fluorescent dye off of a solvent-exposed position of the molecule. Labeling small-molecule ligands with fluorescent dyes is less common for FP assays since the labeling chemistry often results in substantial increases in molecular weight and alterations of molecular properties. In the case of novobiocin, it was hypothesized that such an approach would be feasible since the ligand is relatively large (612 Da), and crystal structures of novobiocin bound to GyrB have revealed the precise binding mode of the ligand, which can be used to identify optimal attachment points. A key advantage of such a system is that the size discrepancy between novobiocin (<1 kDa) and the purified full-length GyrB (92.5 kDa) should be reflected by a large change in polarization values upon binding.
Design and synthesis of Novo-TRX
The crystal structure of novobiocin bound to GyrB (PDB entry 1KIJ) reveals that most of the molecule is deeply buried within the protein active site, with the exception of the phenolic benzamide ring, and the proposed modification was modeled in the active site to confirm solvent accessibility of the attached moiety (see
The novobiocin sodium reacted with formaldehyde and methyl-amine to give a reactive novobiocin methylamine intermediate that could be conjugated to various electrophilic fluorescent dyes (see
Equilibrium binding experiment
Kinetic experiments were performed to determine the time needed for the binding of Novo-TRX to GyrB to reach equilibrium. The experiment examined both the association and dissociation components of the interaction in real time ( Fig. 1 ). To begin, increasing concentrations of GyrB (25, 50, and 100 nM) were mixed with 40 nM Novo-TRX, and the FP signal was monitored every 30 s until equilibrium was reached. The binding of Novo-TRX to GyrB results in an increase in the FP signal over time. As anticipated, the rate of association was faster with higher concentrations of GyrB, as evidenced by the larger initial mP values due to the time resolution of the plate reader. For all reactions, equilibrium was reached by 1000 s (~17 min). To establish that the observed binding was specific, a 125-fold excess (10 µM) of unlabeled novobiocin was added to the samples, and the change in FP signal was measured in real time. The large excess of unlabeled novobiocin was able to compete better than the Novo-TRX for binding to GyrB, and the FP signal decreased over time. As with the association experiments, the new binding equilibrium was reached by roughly 1000 s after the addition of the unlabeled novobiocin. A 1-phase exponential decay equation was used to determine that the average half-life of dissociation was 230 s for the 3 concentrations of GyrB; the association reaction was too fast to determine accurate rates. The ability to monitor binding events in real time in a homogeneous solution is a major advantage of FP over other binding assays that require a separation step to measure the interaction. Binding experiments, including the HTS, were incubated for 60 min before the FP signal was measured to guarantee that equilibrium had been reached in the assay.
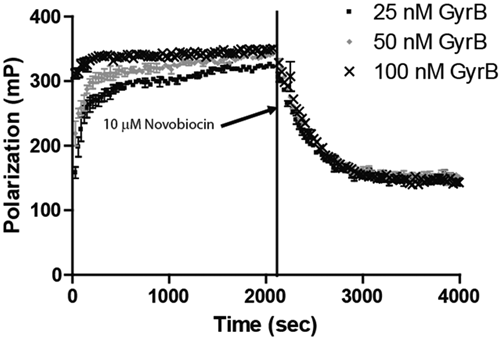
Equilibration studies of the interaction of Novo-TRX and GyrB. Kinetic experiments were performed to determine the time needed for binding of Novo-TRX and GyrB to reach equilibrium. Equal volumes of 2× Novo-TRX were mixed with 2× GyrB to result in a final concentration of 40 nM Novo-TRX and 25, 50, or 100 nM GyrB, and the polarization was measured every 30 s for 70 cycles. The fluorescence polarization (FP) signal (mP) and the error bars represent the data from 4 replicates. To measure the dissociation of Novo-TRX, 125-fold unlabeled novobiocin was added to the Novo-TRX/GyrB samples, resulting in a final concentration of 10 µM. The change in FP was measured every 30 s for 70 cycles. The average FP signal (mP) for 4 replicates was determined, and the error bars represent the standard deviation.
Saturation binding experiment
Experiments were performed to determine the concentration of GyrB that was necessary to reach 100% saturation of binding to Novo-TRX. Preliminary experiments performed with concentrations of Novo-TRX ranging from 1 to 80 nM determined that 20 to 80 nM Novo-TRX resulted in the best window of the assay and Z′ value (see
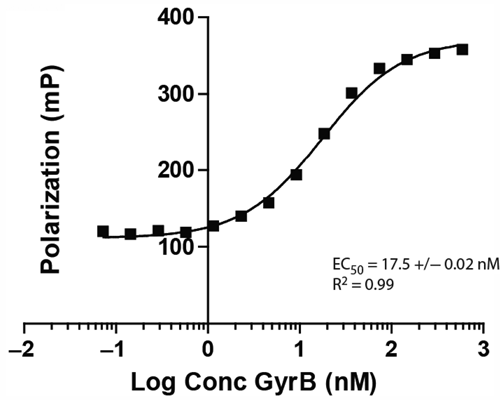
Saturation binding experiments for the interaction of Novo-TRX with GyrB. The homogeneous binding of Novo-TRX to GyrB was measured under equilibrium conditions to determine the amount of GyrB needed to saturate binding. Increasing concentrations (0-2500 nM) of GyrB were incubated with 40 nM Novo-TRX and incubated for 1 h before the binding was measured. The average fluorescence polarization (FP) signal (mP) for 4 replicates was determined, and the error bars represent the standard deviation. To determine the EC50, the data were fit using a sigmoidal dose-response equation.
Solvent susceptibility experiment
Before performing HTS, we tested common solvents and additives for their effect on the binding of Novo-TRX to GyrB using the FP binding assay. The concentration of 40 nM Novo-TRX and 40 nM GyrB was chosen so that binding was roughly 80% saturated. The complex was then incubated with increasing concentrations of DMSO, Triton X-100, Tween-20, and BSA. DMSO, which is commonly used as a solvent for the compounds in a chemical library, had a minimal effect on binding at percentages up to 10% (v/v). Understanding what levels of DMSO could be tolerated by the assay was important, especially in an HTS for small fragments that may have low affinity for the binding site and will need to be tested at higher concentrations (>50 µM). The nonionic detergents, Triton X-100 and Tween-20, also only slightly perturbed binding even at percentages close to 10% ( Fig. 3A ). Additional studies using the detergents in saturation binding experiments demonstrated that the detergents significantly raised the background of the assay (data not shown). It is likely that the level of the signal observed at the highest concentrations of the detergents is equivalent to the background caused by the detergent. It is also possible that the detergents were able to bind to Novo-TRX and increase the FP signal. BSA was well tolerated in the assay up to concentrations around 1 µM; higher concentrations resulted in nonspecific binding ( Fig. 3B ).
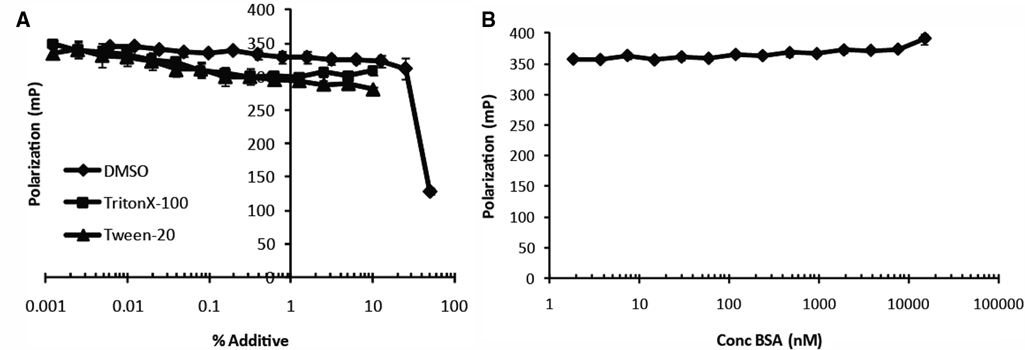
The effect of common additives on the binding of Novo-TRX to GyrB. Common buffer additives were tested for their ability to alter the interaction of 40 nM Novo-TRX with 40 nM GyrB in a homogeneous fluorescence polarization (FP) binding assay. The average FP signal (mP) for 4 replicates was determined, and the error bars represent the standard deviation. (
HTS of a small chemical library
As a proof-of-principle test for the ability of the FP assay to identify inhibitors of the F. tularensis GyrB at the ATP binding domain, the assay was used to screen a small chemical library consisting of 1040 FDA-approved compounds and 80 additional antibiotics ( Fig. 4A - C ). The library was large enough to test the statistical robustness of the assay using multiple liquid handlers that would be required for a large-scale screen. The screen was repeated with a new preparation of GyrB to demonstrate the reproducibility of the assay. The library was chosen because novobiocin was present in multiple locations and would serve as a positive control in the HTS. The compounds were screened at a concentration of 10 µM against a complex of 40 nM Novo-TRX and 40 nM GyrB in 5% DMSO. The Z′ factor, Z factor, and S/N and S/B ratios were calculated as described by Zhang et al. 20 The Z′ factor and Z factor are statistical measures used to determine the quality of the controls and entire data set, respectively. Values between 0.5 and 1.0 indicate that there is significant separation between the high and low signal. The large window of the assay (the distance between the high and low controls), along with the highly consistent data points, resulted in a very robust, reproducible screen (Z′ = 0.8). Both screening efforts had similar Z′ factors, even though the control values are slightly different. The variation in the values between screens is most likely due to differences in purity or activity of the protein preparation used for the screen. In both screens, 8 compounds were identified that decreased the binding of 40 nM Novo-TRX to 40 nM GyrB by greater than 40%: 4 internal novobiocin controls and 4 members of the anthracycline family of cancer chemotherapeutics (doxorubicin, idarubicin, epirubicin, and daunorubicin). This family is known to have anti–topoisomerase II activity, although it is thought that this activity is through DNA intercalation. 23-26
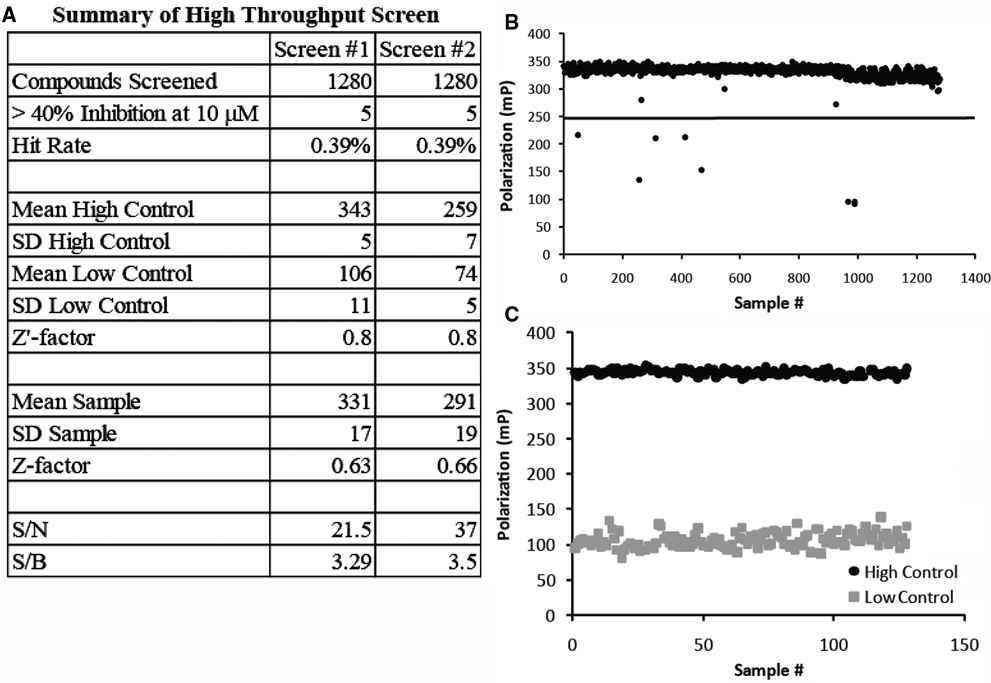
High-throughput screening (HTS) of Food and Drug Administration (FDA)–approved drugs for inhibitors of Novo-TRX binding to GyrB. A small HTS of a library of FDA-approved compounds and 80 additional antibiotics (in triplicate) was repeated twice in a 384-well microplate format to validate the usefulness of the fluorescence polarization (FP) assay in identifying inhibitors of Novo-TRX binding to GyrB. (
Dose-response experiment
The ability of the hits from the HTS to prevent the interaction of Novo-TRX with GyrB was confirmed using a dose-response experiment. Increasing concentrations of the compounds were incubated at room temperature for 1 h with 40 nM Novo-TRX and 40 nM GyrB before the FP signal was measured. The GyrB inhibitor, Vertex-13, 21 was included in the dose-response assay as a positive control to further validate the FP assay with a small-molecule inhibitor that is structurally different from novobiocin. The IC50 was determined by fitting the data using the sigmoidal dose-response equation, as described above ( Fig. 5 ). The IC50 was determined to be 33, 310, 1270, 1580, 1460, and 393 nM for novobiocin, doxorubicin, epirubicin, idarubicin, daunorubicin, and Vertex-13, respectively. Although Vertex-13 is more potent against Staphylococcus aureus and E. coli gyrase than novobiocin, 21 it was found to be approximately 10-fold less potent in the F. tularensis system. This difference in potency could result from structural differences in the ATP binding site of F. tularensis. 22
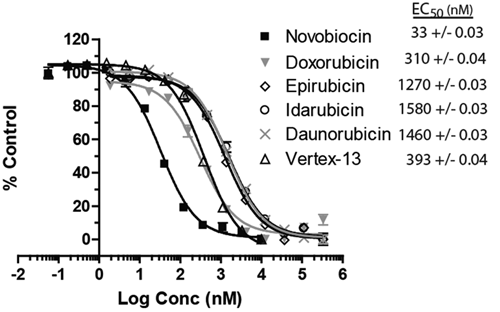
The EC50 of hits from high-throughput screening. Dose-response experiments for the hits from the screen and the small-molecule GyrB inhibitor Vertex-13 were performed using the fluorescence polarization (FP) assay to monitor the inhibition of the binding of 40 nM Novo-TRX to 40 nM GyrB. The average FP signal for each quadruplicate was calculated and normalized to the DMSO control sample. The error bars represent standard deviation of 4 replicates. The EC50 values were determined using the sigmoidal dose-response equation in GraphPad Prism 4.
Gyrase activity assay
The hits were tested in a secondary activity assay to determine if they could prevent the conversion of relaxed plasmid DNA to supercoiled DNA by an E. coli gyrase A2B2 heterotetramer. Initial experiments tested the activity of 10 µM of each compound in preventing the supercoiling of 1.0 µg of relaxed DNA. As anticipated, novobiocin was able to completely prevent supercoiling by the gyrase holoenzyme, whereas none of the anthracycline compounds was able to prevent supercoiling (data not shown). The lack of activity of the anthracyclines was particularly surprising because the compounds are documented to intercalate into DNA and have anti–topoisomerase II activity. 24-26 The ability of the compounds to directly cause DNA supercoiling along with both the gyrase and DNA competing for binding to the anthracyclines, which would result in lower concentrations of compound, most likely resulted in the false-negative activity in the first conditions tested. 27,28 In a second set of experiments, the holoenzyme was incubated with 50 µM of each compound and 0.5 µg of the relaxed DNA for 1 h at 37 °C, and the amount of supercoiling was analyzed by agarose gel electrophoresis and ethidium bromide staining ( Fig. 6 ). DMSO was used as a vehicle control, and presupercoiled DNA alone was loaded as a negative control. A second set of reactions without the gyrase holoenzyme was used to control for supercoiling caused by the compound treatment. Under the second set of conditions, doxorubicin and epirubicin were able to inhibit supercoiling to the level of the no-gyrase control and below the DMSO control. The activity of idarubicin and daunorubicin is less clear because the level of supercoiling caused by the treatment alone is comparable to the level of supercoiling with the gyrase. Although the members of the anthracycline family are known eukaryotic topoisomerase II poisons, they have not previously been reported to directly inhibit the ATP binding domain. Future experiments testing the activity against the eukaryotic topoisomerase and other kinases will be used to determine the selectivity of any hits identified with the FP assay.
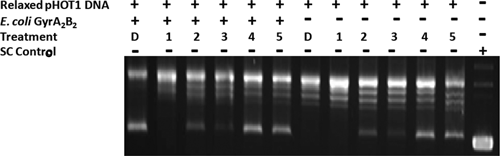
A secondary Gyr activity assay to confirm hits from high-throughput screening (HTS). A secondary assay using the Escherichia coli Gyr A2B2 heterotetramer was used to confirm that the hits from the HTS were able to prevent the supercoiling of 0.5 µg of the relaxed plasmid DNA (pHOT1). Reactions with and without the gyrase heterotetramer were incubated for 1 h with 50 µM of novobiocin (1), doxorubicin (2), epirubicin (3), idarubicin (4), daunorubicin (5), or DMSO (D), and the conversion of relaxed plasmid DNA to supercoiled (SC) DNA was analyzed by electrophoresis on an agarose gel and detected with ethidium bromide staining. Relaxed DNA and SC DNA were included as positive and negative controls, respectively.
Conclusions
We have developed a novel FP assay that specifically measures competition for binding to the ATP binding domain of F. tularensis GyrB. The assay uses a novel binding probe, Novo-TRX, which is composed of a long-wavelength fluorescent dye conjugated to the known ligand novobiocin. This homogeneous, nonradioactive assay has been optimized for screening in a 384-well format and has demonstrated robust assay statistics (Z′ = 0.80). The stability of Novo-TRX has been demonstrated to be suitable for real-time binding kinetics studies and can be used to determine the binding kinetics of potential inhibitors. The high level of conservation in the ATP binding domain suggests that the Novo-TRX probe would be a valuable tool to study the GyrB of any bacterial species. This new methodology has been successfully used to rapidly screen a small library of compounds, and a novel mechanism for the direct inhibition of bacterial gyrase by the anthracyclines was identified and confirmed using a secondary activity assay.
Footnotes
Acknowledgements
The project described was supported by award number U01AI082070 from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.