
Abstract
To facilitate discovery of compounds modulating sphingosine-1-phosphate (S1P) signaling, the authors used high-throughput mass spectrometry technology to measure S1P formation in human whole blood. Since blood contains endogenous sphingosine (SPH) and S1P, mass spectrometry was chosen to detect the conversion of an exogenously added 17-carbon-long variant of sphingosine, C17SPH, into C17S1P. The authors developed procedures to achieve homogeneous mixing of whole blood in 384-well plates and for a method requiring minimal manipulations to extract S1P from blood in 96- and 384-well plates prior to analyses using the RapidFire® mass spectrometry system.
Introduction
S
Standard SphK assays are based on incorporation of radioactively labeled adenosine triphosphate (ATP) into sphingosine or on following conversion of fluorescently or radioactively labeled sphingosine into S1P. 3-5 More recently, mass spectrometry methodology has been applied for precise quantification of bioactive lipids, and methods have been developed for extraction and measurement of S1P from biological samples. 6-9 Here we describe adaptation of RapidFire® mass spectrometry 10,11 to a high-throughput assay measuring S1P formation in human whole blood. Since blood contains endogenous SphK, sphingosine, and S1P, 12,13 we designed a liquid chromatography/mass spectrometry (LC/MS)–based assay that detects the conversion of an exogenously added 17-carbon-long variant of sphingosine, C17SPH, into C17S1P, and we show that the assay is useful for characterization of SphK inhibitors.
Materials and Methods
Reagents and biological material
Fresh sodium heparin–treated human whole blood from healthy, drug-free volunteers was used within 1 to 5 h. To prepare plasma, the blood was pooled and an aliquot was centrifuged at 2000 rpm for 10 min in rotor A-4-62 (Eppendorf model 5810).
High-performance liquid chromatography (HPLC) grade DMSO, methanol, and 40% dimethylamine were from Sigma-Aldrich (St. Louis, MO); acetonitrile and formic acid (88%) were from J. T. Baker (Phillisburg, NJ). D-erythro-sphingosine (d18:1) (C18SPH), D-erythro-sphingosine-1-phosphate (d18:1) (C18S1P), D-erythro-sphingosine (d17:1) (C17SPH), D-erythro-sphingosine-1-phosphate (d17:1) (C17S1P), N,N-dimethyl sphingosine (d18:1) (DMS), 3 and D-erythro-sphingosine-1-phosphate (C20 base) (C20S1P) were from Avanti Polar Lipids (Alabaster, AL). The sphingosine kinase inhibitor, 2-(p-Hydroxyanilino)-4-(p-chlorophenyl) thiazole, HCl (SKI-2), 14 was from EMD Biosciences (San Diego, CA).
Sphingosine and sphingosine-1-phosphate preparation
Chloroform suspensions of C17SPH and DMS were evaporated under nitrogen, dissolved in DMSO (10 mM), and stored at −20°C. Working solutions of C17SPH were prepared in Dulbecco’s modified Eagle’s medium (DMEM)/F12 or RPMI-1640 (Gibco, Grand Island, NY) with 10% heat inactivated fetal bovine serum (HI-FBS) from SAFC Biosciences (St. Louis, MO) and were made fresh before each assay.
C17S1P, C18S1P, and C20S1P were dissolved (1 mg/mL or 2.74 mM, 2.63 mM, and 2.46 mM, respectively) in methanol/water/40% dimethylamine (218:10:2 vol/vol/vol) and stored at −20°C. Working solutions were prepared in acetonitrile and stored at room temperature (RT). C20S1P was added as an internal standard to acetonitrile at 49.2 nM (20 ng/mL; Fig. 2A , B only) or 246 nM (100 ng/mL) to extract the blood.
Blood C17S1P formation assay
Inhibitor compounds were serially diluted in 100% DMSO. Typically, 100 µL of blood was added to a 96-well deep-well assay plate (1.2 mL; #82007-292, VWR, West Chester, PA) containing 1 µL of tested compound in DMSO and mixed for a few seconds with a 96-well, long-tipped, disposable pin tool (#VP-246A, VP Scientific, San Diego, CA) moving freely in the wells while the plate was shaken on a Titer Plate Shaker® (Thermo Scientific, Waltham, MA). After mixing, the plate was incubated at 37°C for 30 min. The reactions were started by the addition of 10 µL/well of 0.1 mM C17SPH substrate in RPMI-1640, 10% HI-FBS. The samples were mixed briefly and then incubated for 8 min at 37°C. Formic acid was added to blood (8.8% final concentration) to stop the SphK reaction and, through acidification, release the protein-bound S1P. This was followed by extraction and protein precipitation with 5 to 7 volumes of acetonitrile containing 246 nM (100 ng/mL) C20S1P internal standard (IS). Combination of the acid and acetonitrile treatments into 1 step was avoided as it resulted in increased variability in data (data not shown). The precipitated solution was mixed on the plate shaker while manually holding the pin tool in the sample wells. Mixing continued for ~15 s on setting 2, until the supernatant was a uniform dark brown color. After precipitation, plates were sealed with adhesive foil and centrifuged (4000 rpm, 10 min, RT). A 25-µL aliquot of the supernatant was diluted 8- to 10-fold into 35% acetonitrile. Aliquots (100 µL) of diluted sample were transferred to a 96-well plate (#5042-1386, Agilent, Santa Clara, CA). The sample plates were heat sealed with AB-3738 Easy Pierce Heat Sealing Foil® (Thermo Scientific), then stored at −80°C and shipped on dry ice to BIOCIUS Life Sciences for RapidFire® mass spectrometry (RF-MS) analysis. Plates remained sealed throughout the RF-MS analysis to prevent evaporation and analyte precipitation.
Extraction efficiency experiment
One hundred µL/well of blood with 1% DMSO was incubated at 37°C for 30 min in 96-deep-well plates. Dilution series of S1P standards were prepared in acetonitrile, and 6 µL was added to the blood/DMSO and incubated at 37°C for 8 min before extraction. S1P was extracted by treatment with formic acid (17.6% final concentration) and acetonitrile/C20S1P (7 volumes). The standards were also diluted into vehicle consisting of formic acid and acetonitrile/C20S1P with phosphate-buffered saline (PBS) instead of blood. Following treatment, the supernatant was diluted into 35% acetonitrile (1:10 vol/vol) and analyzed by RF-MS.
RF-MS methods
Briefly, the RF-MS platform is a fully automated, online, microfluidic sample preparation system. 10,11 Ten µL of sample is aspirated directly from 96- or 384-well assay plates and loaded onto the RF-MS microscale solid-phase extraction cartridge. The salts, buffers, cofactors, and so on are washed through the cartridge while the analytes of interest are retained. The purified analytes are then eluted off of the cartridge directly onto the mass spectrometer for analysis.
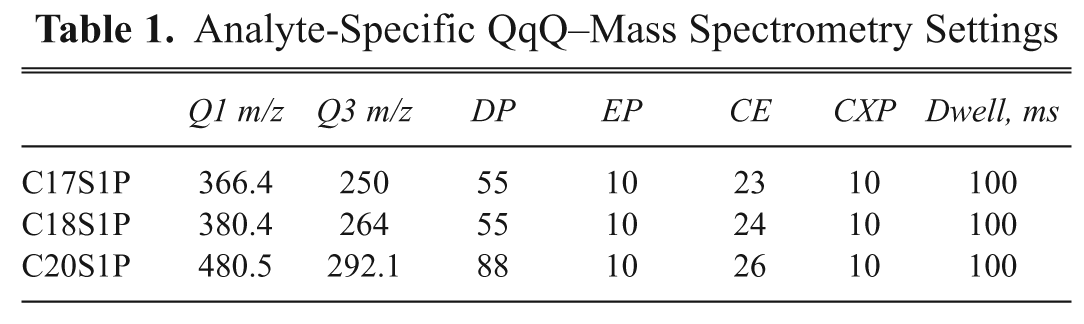
C17S1P, C18S1P, and C20S1P were monitored on a Sciex API-4000 triple quadrupole mass spectrometer in the positive electron spectroscopic imaging (ESI) mode using the conditions outlined in Table 1 . Samples were delivered from assay plates onto the RF-MS microscale solid-phase C4 extraction cartridge (BioTrove column A) to remove nonvolatile assay components with HPLC grade ddH20 containing 0.1% trifluoroacetic acid (TFA) in a 3-s wash cycle. All analytes were coeluted into the mass spectrometer in a 3-s elution cycle using 80% acetonitrile containing 0.1% TFA. The chromatography system produced baseline resolved peaks with widths of ~2 s. The total cycle time, including wash, elution, and reequilibration of the cartridge, was ~6.5 s per sample with analysis times of ~42 min per 384-well plate.
Analyte-Specific QqQ–Mass Spectrometry Settings
384-well IC50 blood assay procedure
Twenty µL/well of blood was added to the 384-well compound assay plate (#781280, Greiner, Monroe, NC) containing 250 nL/well of diluted compound in DMSO that was dispensed beforehand using a Cartesian Hummingbird® nanoliter liquid dispenser (Cartesian Technologies, Irvine, CA). SKI-2 at a final concentration of 100 µM and DMSO served as the 100% and 0% inhibition controls. The blood was mixed with compounds either by Biomek FX® using AP384 P30 pipette tips (Beckman Coulter, Brea, CA; 20 µL, 5 repetitions) or by inserting a 384-well pin tool (#VP-248, VP Scientific) into the wells and shaking for several seconds on a plate shaker. The plate was covered and incubated for 30 min at 37°C. The reactions were started by the addition of 5 µL/well of 0.1 mM C17SPH in RPMI, 10% FBS. The samples were mixed, either by pin tool/plate shaker or Biomek FX®, and then incubated for 8 min at 37°C. Then, 6 µL/well of 33% formic acid was added by Biomek FX®. The samples were immediately stirred manually with a pin tool for ~10 s, and then 70 µL/well of acetonitrile/C20S1P was added using a Multidrop® (Thermo Scientific) liquid handler. Precipitated samples were mixed by pin tool/plate shaker for ~15 s, as described. Following centrifugation, a 10-µL aliquot of supernatant (Biomek FX®) was added to a 384-well plate containing 90 µL/well of 35% acetonitrile. The dilution plate samples were mixed by pin tool/plate shaker for several seconds or by Biomek FX®. The Biomek FX® required 10 to 15 repetitions of aspirating 30 µL from the bottom of the well and dispensing it to the top to mix the different densities of extract and diluent. A 50-µL aliquot of diluted sample was prepared by Biomek FX® for RF-MS and was heat sealed and frozen. Samples were analyzed for C17S1P, C18S1P, and C20S1P. The C17S1P data were normalized to C18S1P native analyte. The percent of C17S1P formation was calculated as (sample AUC value/vehicle AUC value) × 100. IC50 values were calculated by nonlinear fit–sigmoidal dose response using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA).
Results and Discussion
Exogenously added sphingosine is efficiently taken up by cells in culture and/or by blood cells and converted by the intracellular sphingosine kinases into sphingosine-1-phosphate. 3,13 To begin to quantify the conversion of exogenously added C17SPH into C17S1P by blood using RF-MS spectroscopy, we first determined the efficiency of S1P extraction from blood. Various amounts of C17S1P or C18S1P were added to blood, extracted by treatment with formic acid and acetonitrile, and quantified by RF-MS as described in the Materials and Methods. In comparison to a similar extraction regimen in which the analytes were added to PBS, the recovery of C17S1P and C18S1P from blood was 27% and 22%, respectively ( Fig. 1 ). As shown, extraction was linear with added S1P in the range of concentrations used ( Fig. 1 ).
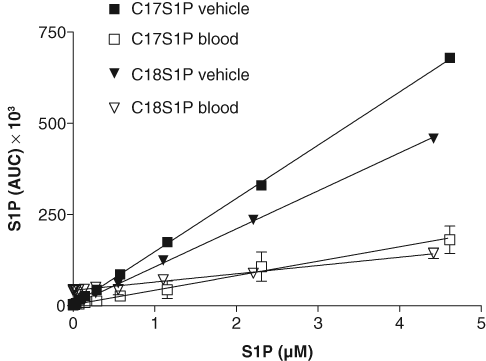
Recovery of exogenously added sphingosine-1-phosphate from blood. Serially diluted C17S1P (final concentration range 4.614-0.018 µM) and C18S1P (4.415-0.017 µM) were added to blood or phosphate-buffered saline (vehicle), extracted with formic acid/acetonitrile treatment, and quantified by mass spectrometry. The data points are the averages of the RapidFire® mass spectrometry area under the curve (AUC) peak values (see Fig. 3C for an example). Percent recovery was determined by comparing linear regression slopes of blood and vehicle extracts (% Recovery = slope for blood × 100/slope for vehicle) using GraphPad Prism software.
We next determined the kinetics of conversion of C17SPH into C17S1P in human plasma and blood using varying concentrations of the substrate ( Fig. 2A , B ). In plasma, the reaction was linear for at least 15 min, and the initial reaction rate appeared to be saturated at 5 µM C17SPH. In blood, the initial conversion rate was much higher, and the reactions were linear for about 5 to 10 min followed by plateaus with a slight decreasing slope as the time approached 45 min. This is consistent with observations that the specific activity of sphingosine kinase is significantly higher in mouse blood compared to plasma. 13 To determine the reason for the C17S1P plateaus in whole blood, we did a second time course experiment with 20 µM C17SPH substrate added to blood at 0 and 25 min of incubation ( Fig. 2C ). After the second addition of substrate, there was an increase in the release of C17S1P similar to that seen after the first addition. This indicates that the endogenous SphK remains active 25 min after the start of the experiment, and the observed plateaus in formation of C17S1P in the whole blood ( Fig. 2B ) were due to depletion of substrate and not inactivation of the enzyme. The percent conversion of the 20-µM C17SPH substrate at 7 and 10 min was 35.3% and 44.1%, respectively. The levels of endogenous C18S1P were not affected by the addition of C17SPH substrate or by incubation at 37°C during the 45 min of the assay. This is consistent with reports that, due to the lack of S1P lyase and low activity of S1P phosphatases in platlets and red blood cells (RBCs), S1P is relatively stable in ex vivo blood. 12,15,16 From these data, we chose 20 µM C17SPH with an 8-min incubation at 37°C for testing the activity of sphingosine kinase inhibitors in both the 96- and the 384-well formats of the blood assay.
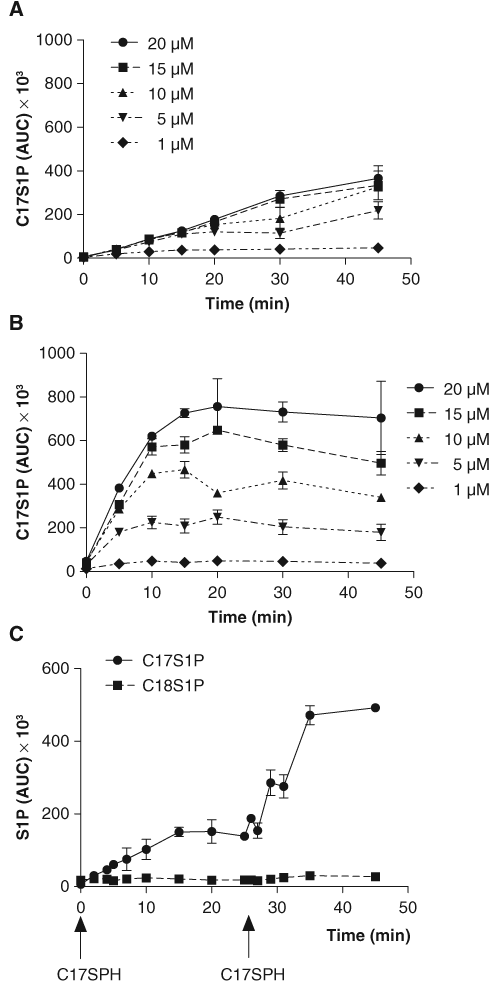
Conversion of C17SPH to C17S1P in (
Figure 3 shows dose-response curves of the inhibition of C17SPH conversion to C17S1P by standard SphK1 inhibitors, SKI-2 and DMS, in the 96- and 384-well assays. The C18S1P concentrations remained stable (data not shown), consistent with the pattern observed in Figure 2C . The fact that C18S1P levels remained stable across all samples, regardless of treatment, provided a control for S1P extraction efficiency throughout the plate. To monitor variations in loading volume onto the RF-MS, we also included C20S1P as an internal standard in the acetonitrile used for extraction. However, we found that normalizing the C17S1P data to C18S1P was more useful, as S1P extraction proved to be the step with the highest potential for variability. The IC50 values obtained in both formats of the blood assay were very similar and were 2- to 4-fold higher than the values obtained for those inhibitors in reactions run with purified enzyme. 5,14 Such a right shift in the inhibitor potency in the blood assay is commonly observed and results from sequestration of the compound due to, for example, protein binding or decreased ability to penetrate cell membranes. The Z′ values 17 calculated from control samples run in the IC50 experiments were 0.79 for the 96-well format and 0.55 for the 384-well format, confirming that the assay is suitable for high-throughput applications ( Fig. 3A , B ).
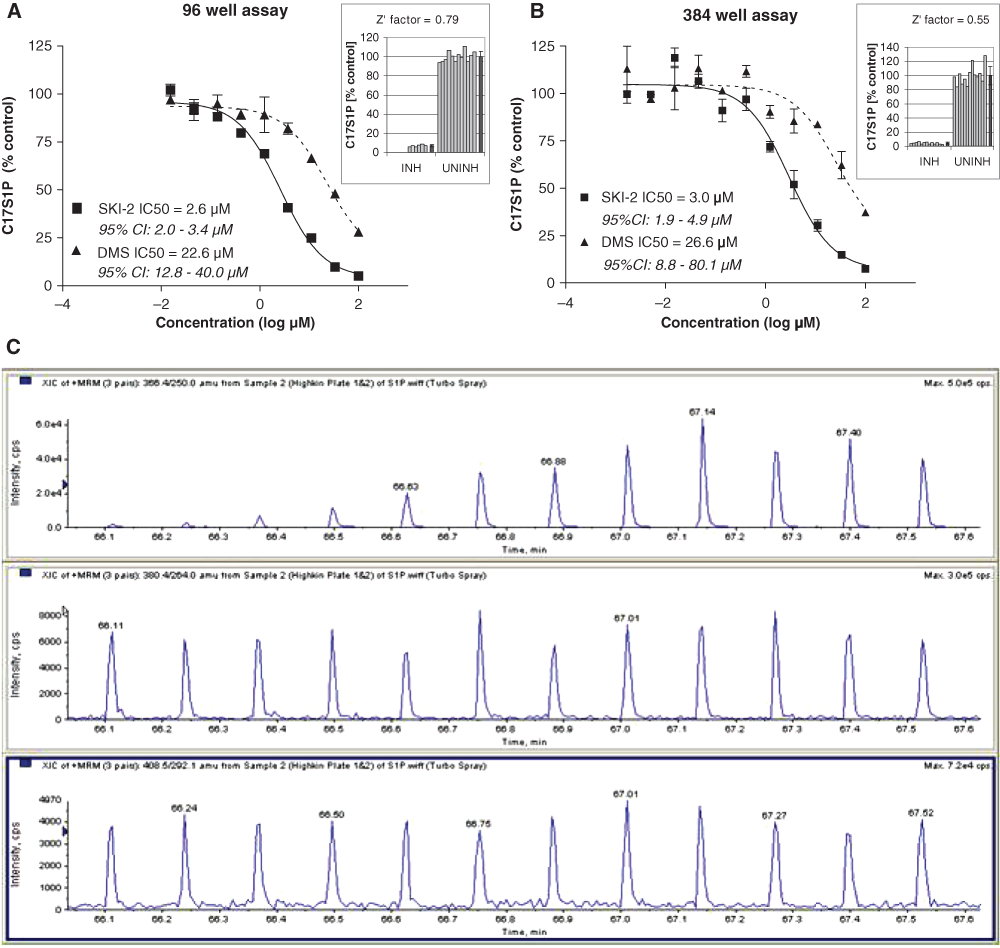
Inhibition of C17S1P formation in human whole blood by sphingosine kinase inhibitors, DMS and SKI-2, measured by RapidFire® mass spectrometry (RF-MS) in the (
The dose-response curves from SKI-2 and DMS show that the assay can differentiate between inhibitors with different potencies against SphK in blood. The assay does not distinguish between the 2 forms of SphK (SphK1 is the predominant activity in blood). 4,13
Adapting whole-blood assays to 384-well plates is complicated by difficulty in achieving homogeneous mixing due to blood’s viscosity and by factors related to the changed dimensions of the wells in 384-well plates compared to 96-well plates. 18 As described, 18 blood was mixed in 384-well plates with disposable pin tools and a tabletop plate shaker or by aspirating sample from the bottom of the well and dispensing it to the top using a Biomek FX®. Mixing with a pin tool was critical once the blood protein had been precipitated. This step can be performed with the pin tool held manually or preferably using a jig and orbital plate shaker system, such as the VP-177 STIR® (VP Scientific), which holds the pin tool at a set height while rotating the plate in a 2-mm amplitude.
Conclusions
We describe a rapid ex vivo IC50 blood assay in 96- and 384-well plates, which can determine relative potencies of sphingosine kinase inhibitors. Whole-blood assays are helpful in identifying inhibitors that not only inhibit enzymatic activity but can also reach the target enzyme in the tissue and are not neutralized by protein binding or inability to penetrate cells and can serve as a better predictor of dosing concentrations for in vivo models than enzyme and cell assays. 19
RapidFire® RF-MS was chosen to measure S1P because its throughput rates made mass spectrometric analysis feasible for multiple 384-well plate assays. Also, its ability to remove DMSO, salts, and buffers incompatible with mass spectrometry simplified the preparation of blood extracts. Although RF-MS assays have been previously described 10,11 for complex matrixes such as diluted cell lysates, liver homogenates, or FBS up to 15%, 10 our assay expands the use of this technology to whole blood in 384-well plates.
Footnotes
Acknowledgements
We thank Michael Prinsen for suggesting RF-MS; Becky Hood, Jian Zhang, and Sheri Bonar for training in automation equipment; Rebecca Johnston, Daniel Krupa, and Martin Meier for compound plate preparation; and Mark Obukowicz, Rita Huff, and Arthur Wittwer for scientific discussions.