
Abstract
Sphingosine kinase 1 (SPHK1) regulates cell proliferation and survival by converting sphingosine to the signaling mediator sphingosine 1-phosphate (S1P). SPHK1 is widely overexpressed in most cancers, promoting tumor progression and is associated with clinical prognosis. Numerous studies have explored SPHK1 as a promising target for cancer therapy. However, due to insufficient knowledge of SPHK1 oncogenic mechanisms, its inhibitors’ therapeutic potential in preventing and treating cancer still needs further investigation. In this review, we summarized the metabolic balance regulated by the SPHK1/S1P signaling pathway and highlighted the oncogenic mechanisms of SPHK1 via the upregulation of autophagy, proliferation, and survival, migration, angiogenesis and inflammation, and inhibition of apoptosis. Drug candidates targeting SPHK1 were also discussed at the end. This review provides new insights into the oncogenic effect of SPHK1 and sheds light on the future direction for targeting SPHK1 as cancer therapy.
Keywords
Introduction
Sphingosine kinase 1(SPHK1/SK1/SPK1) mediates the conversion of sphingosine (Sph) to sphingosine-1-phosphate (S1P), a pivotal sphingolipid signaling mediator involved in a wide variety of cellular processes, including cell growth, survival, differentiation, and motility. 1,2 The activation of SPHK1 requires 2 events: phosphorylated by extracellular signal-regulated kinase 1/2 (ERK1/2) and translocation to plasmalemma. 3,4 Two mammalian isoenzymes have been identified, SPHK1 and SPHK2. 5,6 SPHK1 localizes predominantly in the cytosol as it contains a functional nuclear export sequence, whereas SPHK2 locates both in the nuclei and the cytoplasm, indicating their distinct biological roles. 7 Since the first human SPHK1 structure was illustrated, 8 significant advances have been made in understanding the composition and function of SPHK1. 9 Human SPHK1 gene is localized to 17q25.2, and its protein products have 3 splice isoforms, SPHK1a, SPHK1b, and SPHK1c. SPHK1a is primarily involved in the extracellular signaling transduction, while SPHK1b and SPHK1c are mainly anchored on the cell membrane. 10 The structure of SPHK1 adopts a 2-domain architecture that comprises 9 α helices, 17 β strands, and a 310-helix (Figure 1). 8,11 Each motif has its specific binding sites associated closely with its functions, such as active sites, nucleotide-binding sites, calcium/calmodulin(Ca2+/CaM) coupling sites, magnesium ion combining sites, lipid associating sites, phosphorylation/dephosphorylation sites, etc. (Table 1). Although the structure has been characterized, 8,9 molecular mechanisms of SPHK1 functions, such as its translocation activation, and interactions between 3 various isoforms and other molecules, are poorly understood. SPHK1 is a perplexing kinase with pivotal roles in various cellular processes (Figure 2). Cumulating evidence has revealed that SPHK1 is upregulated in cancer cells and closely correlated with tumors progression 16 -19 (Table 2), and high expression level or kinase activity of SPHK1 is associated with a poor prognosis in several types of cancers. Recently, oncogenic mechanisms of SPHK1 have been identified in multiple aspects, including tumor growth, tumor migration and angiogenesis, and so on. This review introduced the SPHK1/S1P signaling pathway and discussed the potential mechanism of SPHK1/S1P signaling in tumor progression. Besides, inhibitors of SPHK1 with therapeutic potentials were also summarized.

The basic structure of SPHK1. SPHK1 consists of NTD and CTD, 5 conserved motifs, C1-C5. C1-C3 involved in NTD, while C4-C5 in CTD (C1:19-36aa, C2:72-96aa, C3108-119aa, C4:165-198aa, C5:338-343aa). Every domain has specific motifs, 9 α helices, 17 β strands, and a 310-helix.

The schematic of the SPHK1/S1P signaling pathway. SPHK1 catalyzes the formation of S1P from Sph and is pivotal regulators of the balance between Cer, Sph, and S1P. The gene expression of SPHK1 is regulated by a transcription factor (TF), such as transcription factor specificity protein 1 (Sp1), AP2, E2F transcription factor, hypoxia inducible factors (HIF), LIM-domain-only protein 2 (LMO2). After expression in the cytoplasm, SPHK1 is activated by ERK1/2, resulting in its phosphorylation and translocation to the cell membrane; CIB1 participates in this process. When SPHK1 is fitted on the cell membrane, it will catalyze the production of S1P, and the yielded S1P is secreted to the extracellular domain via the S1P channels (ABCA1/B1/C1, Spns2), and then the extracellular S1P engaged with the receptors (S1PR1-5) of other cells or itself cell membrane. Owning to S1PR is a G-protein coupled receptor (GPCR) that could transmit extracellular signaling into intracellular second message S1P, initiates the classical GPCR signaling pathway, and elicits serious effects, including cell metabolism, proliferation, migration, angiogenesis, autophagy, apoptosis, inflammation, etc.
The Specific Binding Sites of SPHK1.a

a SPHK1 contains multiple sites, such as active site (which is essential for the function of this enzyme), nucleotide-binding sites (bind ATP, as a substrate to phosphorylate Sph), Ca2+/CaM binding sites (bind Ca2+/CaM or act as a cellular calcium sensor for SPHK1), magnesium ion binding sites (coordinate to the γ-phosphate group of ATP and the pyrophosphate moiety), lipid biding sites (bind with Sph and act like a lipid gate controlling the in-and-out of lipid substrate and product), ERK1/2 and PKC phosphorylation sites (phosphorylate SPHK1) and PP2A-B’α dephosphorylation site (dephosphorylate SPHK1).
The Expression and Potential Mechanism of SPHK1 in Human Cancers.a

a SPHK1 is elevated in many tumors, mostly more than twice that of normal cells (non-tumor cells). And lots of associated molecules participate in the process of cancer. EMT: epithelial-mesenchymal transition; 67LR: 67-kDa laminin receptors; ER: estrogen receptor; TRAF2: TNF-receptor-associated factor.
SPHK1/S1P/S1PR Signaling Pathway
SPHK1 is one of the versatile kinases that catalyze the synthesis of S1P, and it performs an increasingly essential role in regulating the metabolic balance of sphingolipids such as ceramide (Cer), sphingosine (Sph), and S1P. 58 Cer plays a vital role in apoptosis, cell senescence, differentiation, and cellular stress responses, 58 -60 and Sph is an anti-growth signaling molecule. 61 In contrast, S1P promotes cell proliferation, survival and inhibits apoptosis. 62 -64 The SPHK1/S1P signaling plays a crucial role in inflammation, cell migration, and vascular development, 16 which has been inextricably linked to tumorigenesis. SPHK1 also acts on D-erythro-sphingosine in vitro 8 and, to a lesser extent, sphinganine 65 through phosphorylation. Moreover, SPHK1 has serine acetyltransferase activity on cyclooxygenase 2 (COX2) in an acetyl-CoA dependent manner, which contributes to pathogenesis in a model of Alzheimer’s disease (AD).66
SPHK1 has been identified as an important regulator in regulating extracellular and intracellular S1P levels (Figure 2). In normal conditions, SPHK1 is a predominantly cytosolic enzyme. 9 The expression of SPHK1 is regulated by the transcription factors (TF), such as transcription factor specificity protein 1 (Sp1), AP2, E2F transcription factor, hypoxia-inducible factors (HIF), LIM-domain-only protein 2 (LMO2) 4 (Figure 2). When exposure to several stimuli, such as growth factors, cytokines, and hormones, SPHK1 is activated and translocated to the cell membrane, catalyzes the production of S1P, thereby raising S1P contents. 67 -69 As a second messager, S1P is a biologically active lipid catalyzed by either SPHK1 or SPHK2, which can be released from cells via specific channels such as ATP-binding cassette (ABC-A1/B1/C1), major facilitator superfamily transporter 2b (Mfsd2b), and spinster homolog 2 (Spns2) 70 -72 to target to the G protein-coupled receptor family (S1PR1-5) proteins. 10 In the cytoplasm, S1P can also couple with specific partners, such as histone deacetylases (HDAC1/2), human telomerase reverse transcriptase (hTERT), TNF-receptor-associated factor(TRAF2) 73 -75 and prohibitin 2 (PHB2), 76 to trigger a variety of cellular responses.
On the other hand, the activity of SPHK1 is also crucial for the clearance of sphingolipids. Once S1P comes into being, S1P is hydrolyzed by S1P lyase in an irreversible reaction to yield hexadecenal and phosphoethanolamine (Figure 2), which is an important exit of sphingolipid metabolic cycle. 58 The oncogenic signaling of SPHK1 is dependent on its phosphorylation activation at Ser225 by ERK1/2 and shifts from the cytoplasm to the plasma membrane. 3,77 ERK1/2 not only stimulates the catalytic activity but also is necessary for SPHK1 plasma membrane trafficking. 78 Finally, inactivation of SPHK1 is dephosphorylated by specific protein phosphatase, such as protein phosphatase 2A (PP2A, Table 1). It has been demonstrated that B’α (B56α/PR61α/PPP2R5A), a regulatory subunit of PP2A, interacts with the c-terminus of SPHK1, then decrease SPHK1 phosphorylation. 14 SPHK1 is degraded through either the proteasome or lysosomal pathways in response to several stimuli, such as DNA damage, tumor protein 53 (TP53), tumor necrosis factor α (TNF-α), and even its inhibitors. 4
Nevertheless, the SPHK1 membrane localization mechanism is still unclear, which might be associated with the activation of ERK1/2 or other partners like Ca2+/CaM and PKC. 79,80 The activation of SPHK1 and its subsequent translocation to the plasma membrane was observed in response to the PKC activator, PMA (Phorbol 12-myristate 13-acetate). 80 Although SPHK1 contains 4 putative PKC phosphorylation sites (shown in Table 1), purified PKC does not affect SPHK1 activity in vitro, 81 indicating an indirect mechanism of PKC on SPHK1 activation. As for Ca2+, chelation of intracellular Ca2+ inhibits S1P production, whereas increasing intracellular Ca2+ enhances S1P formation. 82 It has been shown that Ca2+ plays an essential role for CaM in the Ca2+-dependent translocation of SPHK1 to the plasma membrane. 14 However, although several putative CaM-binding sites have been identified by sequence analysis of SPHK1 (Table 1), direct activation of SPHK1 by Ca2+ has not been demonstrated so far. Besides, several pieces of evidence demonstrated that the translocation of SPHK1 to the cell membrane is mediated by calcium and integrin-binding protein (CIB1) through its Ca2+ myristoyl switch function, and the SPHK1 signal transduction may be achieved through the Ras pathway. 83 -85 To a large extent, CIB1 is associated with the perplexing structure of SPHK1, 9 while CIB2 has opposite actions. 32 Based on these findings, Ca2+ may perform its functions by binding their partner, especially CaM and CIB1 (Figure 2).
Interestingly, Adams et al. proposed that the translocation of SPHK1 to the plasma membrane also attributes to SPHK1 dimerization. 9,86 Specifically, SPHK1 forms dimers and interacts with plasma membranes through a single contiguous interface that would elongate the interface and strengthen the SPHK1-phospholipids interaction. 4 However, the kinetics and dynamics underlying SPHK1 dimerization and how that would affect membrane binding and function in cells need to be further investigated.
Of note, cytokines such as TNF-α, interleukin-1 (IL-1), or growth factors have been reported to regulate the SPHK1/S1P signaling pathway, affecting the formation of S1P. 18,87 Similarly, hormones are also fundamental stimulation signals, impacting SPHK1, and S1P metabolism, thus controlling cell life activities. 88 After activation, SPHK1 promotes tumorigenesis by modulating various processes, such as apoptosis, autophagy, proliferation, migration, invasiveness, angiogenesis, and inflammation (Table 2). As shown in Figure 2, these diversified processes benefit from the significant position of SPHK1 in the SPHK1/S1P signaling pathway. 30,44,89
The Potential Oncogenic Mechanism of SPHK1
SPHK1/S1P axis is implicated in numerous pathophysiological conditions and diseases, such as deafness, hepatitis, diabetes, obesity, atherosclerosis, osteoporosis, Alzheimer’s disease, multiple sclerosis, and even cancer. 90 SPHK1 is widely upregulated across a diverse range of human cancers (Table 2), such as breast cancer, lung cancer, uterine cancer, ovarian cancer, gastric cancer, kidney cancer, liver cancer, prostate cancer, colorectal cancer, small bowel cancer, chronic myeloid leukemia, glioblastoma, and lymphoma. 19,51,91,92 After being activated by phosphorylation, SPHK1 accelerates the synthesis of S1P on the cell membrane. Then the generated S1P as a second messenger, in an autocrine or paracrine manner, engages with S1PRs, to evoke a range of biological functions, such as apoptosis, autophagy, proliferation, migration, invasion, angiogenesis, and inflammation.
Effect of SPHK1 on autophagy and apoptosis
Recent studies have shown that SPHK1 is involved in autophagy and apoptosis. Dysregulation of autophagy was associated with various human disorders, such as neurodegenerative diseases, obesity, infectious diseases, cardiomyopathy, type 2 diabetes, and cancer, 93,94 while apoptosis also plays vital roles in the pathogenesis of these diseases. 95 Hence, both autophagy and apoptosis have been recognized as promising therapeutic targets. In particular, it is proposed that SPHK1 promotes tumorigenesis by simultaneously upregulating autophagy and suppressing apoptosis (Figure 3).

SPHK1 coordinates autophagy and apoptosis. A, The relationship between SPHK and autophagy. On the one hand, autophagy is a self-protection process that provides sufficient energy supply for cell growth by recycling useless raw materials; on the other hand, cancer cell takes advantage of autophagy for tumorigenesis by some mechanisms, such as cell survival, EMT and protecting cell under starvation. SPHK1 is a potential target that inhibiting SPHK1 can induce autophagy-induced death via some special mechanism, for example, TP53. B, The role of SPHK1 in apoptosis. SPHK1 performs an anti-apoptosis effect through PI3K/Akt/NF-κB(or only PI3K/Akt), Akt/FoxO3a/Bim, or RTKs. Targeting SPHK to inducing apoptosis has been a focus on cancer therapy. Many inhibitors can induce apoptosis, including SK1-Ⅰ/Ⅱ ˎ Ski/ DHS ˎ FTY720 ˎ CB5468139 ˎ PF-543 ˎ Safingol, and so on (Table 3). To sum up, there are 4 ways to induce apoptosis by inhibiting SPHK1: promoting hydrolysis, inhibiting translocation to plasmalemma, regulatory RNA, and even a direct inhibition of inhibitor. C, The role of SPHK1 between autophagy and apoptosis; cancer therapy targeting SPHK1. In principle, the relationship between autophagy and apoptosis is antagonistic. But SPHK1 correlates autophagy and apoptosis, and targeting SPHK1 could induce cell death via autophagy and apoptosis.
Autophagy is regulated by more than 30 autophagy-related genes (ATG) and requires functional late endosomes and lysosomes to participate. 96,97 It is unraveled that SK1-I (a specific SPHK1 inhibitor, Table 3) inhibits SPHK1 activity, promotes the fusion of endosomal membranes to accumulate dysfunctional enlarged late endosomes, and blocks autophagy flux. 98 Whereas inhibition of SPHK1 by SK1-I greatly increases autophagic flux and induces TP53-dependent cell death mediated by the autophagic regulators BECN1 and ATG5. 17 In neuronal cells, overexpression of SPHK1 enhances the formation of the pre-autophagocytic Beclin1-positive structure and strengthens autophagy flux, whereas S1PPase and S1PLase have opposite effects on regulating neuronal autophagy. 99 Interestingly, it has been reported that SPHK1 accelerates lysosomal degradation of CDH1 (cadherin family members) to induce epithelial-mesenchymal transition (EMT), which depended on TRAF2-mediated autophagy activation. 100 These findings have revealed a novel mechanism responsible for regulating the EMT via SPHK1/TRAF2/BECN1/CDH1 signal cascades in hepatocellular carcinoma (HCC) cells. 100 MiR506-3p has been demonstrated to act as a tumor suppressor through MET initiation and autophagy inhibition, which can be reversed by SPHK1overexprssion. 55 SPHK1 expression and activity were significantly augmented by TGF-β1 stimulation, resulted in increased expression of autophagy-related genes ATG5/12 and Beclin 1 and subsequent autophagy induction. 101 Moreover, SPHK1 could activate the ERK pathway in colon cancer, 45 which also elicits autophagy. In MDCK cells with high expression of K-Ras, S1P and S1PR2 receptors are down-regulated by K-Ras, while SPHK1 and S1PPase were not affected. The lack of S1P was linked with increased autophagy. 102 It is noteworthy that SPHK1 may also interact with other biologically active substances such as Cer and S1P. 103 It is, therefore, a promising approach to investigate the regulation of sphingolipids (S1P, Cer, and SPHK1 were included) on autophagy and cell death (Figure 3C).
The Potential SPHK1 Inhibitors.a
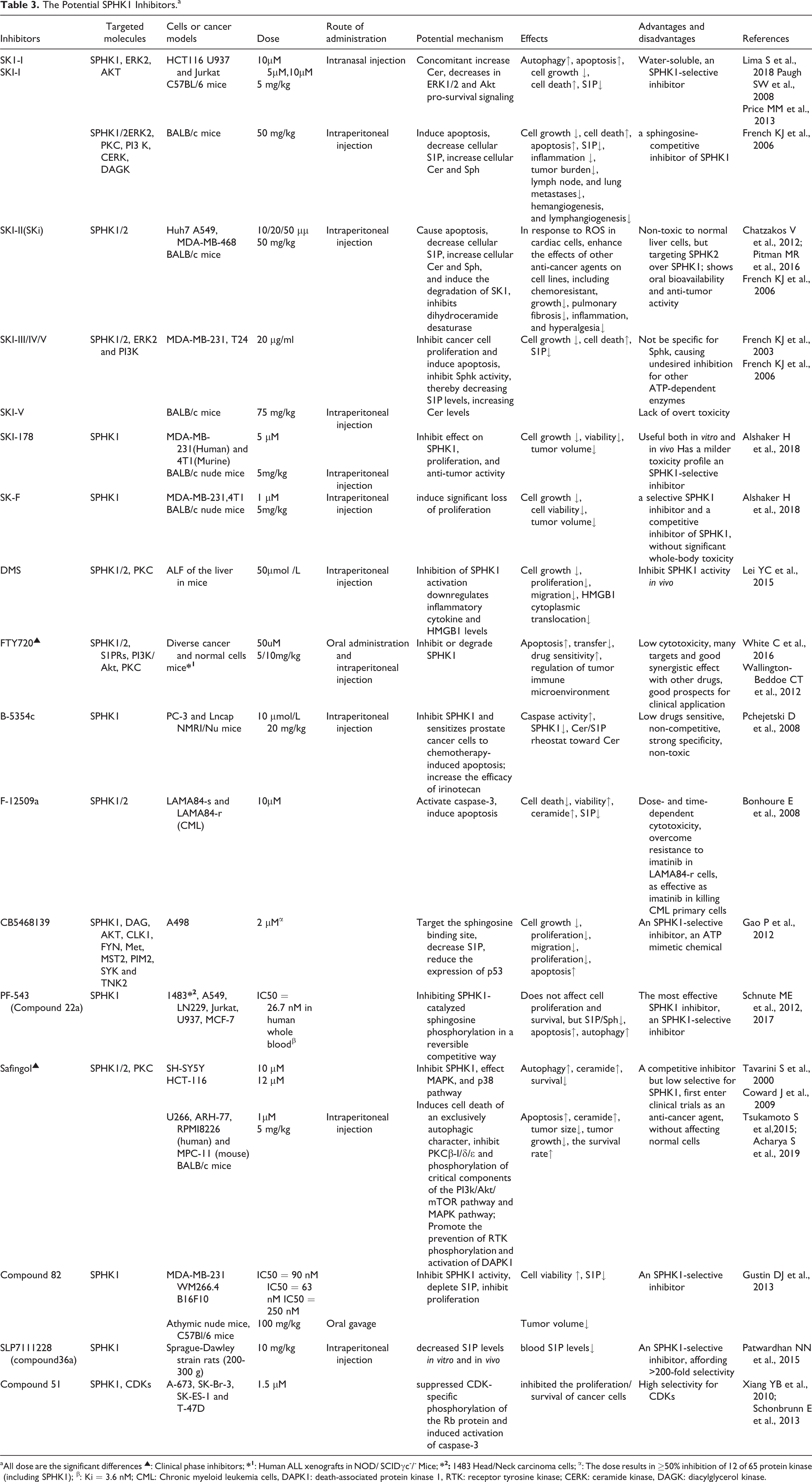
a All dose are the significant differences ▴: Clinical phase inhibitors;
Interestingly, the correlation between autophagy and SPHK1 varies in different tumor types and stages. Overexpression of SPHK1 induces autophagy in neurons and protects against starvation in star glial cells. 104 SPHK1 promotes autophagy in neurons by translocating to endocytic and autophagic vesicles, while in SH-SY5Y neuroblastoma cells, SPHK1 exclusively locates on the endosomal membrane. 105 Autophagy is a double-edged sword in tumor biology. Previous studies demonstrated that autophagy inhibits tumorigenesis in the early stage, while more advanced cancer cells utilize autophagy as a pro-survival tool to enhance tumor growth. 106 -108 SPHK1 knock-out protects TP53 -/- and TP53 +/- mice from developing thymic lymphoma, 109 suggesting that SPHK1 is required for spontaneous tumorigenesis in TP53 -/- mice. Thus, p53 is functionally associated with SPHK1, and SPHK1 could be a downstream target of p53. Furthermore, SPHK1-induced autophagy promotes tumor cell survival, induces EMT, and protects against starvation. These processes are strongly associated with tumorigenesis. Besides, SPHK1 inhibition could induce autophagic cell death, a novel anti-cancer strategy (Figure 3A/C).
SPHK1 also plays a vital role in apoptosis. Inhibition of SPHK1 results in lower S1P levels and apoptosis of prostate cancer cells. 110 Similar results have been reproduced in other types of cancer (Table 2). Recent reports show that p53-mediated caspase-2 activation is required for SPHK1 proteolysis. Caspase-2 activation is downstream of the checkpoint kinase 1 (CHK1, a serine/threonine-protein kinase) inhibitory pathway in TP53-mutated cells, and inhibition of CHK1 leads to caspase-2 activation and apoptosis. 111 The aforementioned p53 activation by inhibition of SPHK1 leads to pro-apoptotic BCL2 family members’ upregulation, including BAD, BAK1, and BID. 17 CIB2 blocks the translocation of SPHK1 to the plasma membrane and impairs its subsequent signaling, including induction of apoptosis by TNF-α and limiting Ras-induced tumorigenesis. 32 In contrast, CIB1 mediates the translocation of SPHK1 to cell membranes. 83 -85 but whether CIB1 participates in apoptosis is unclear.
A recent study in non-small cell lung cancer (NSCLC) has revealed that the upregulation of SPHK1 boosts the PI3K/AKT/NF-kB pathway, and suppressing this pathway abolishes the anti-apoptotic effect of SPHK1. 112 However, neither SPHK1 nor SPHK2 regulates NF-kB activation. 113 Suppression of SPHK1 induces apoptosis in glioblastoma cells and limits tumor growth. SK1-I specifically restrains phosphorylation and activation of AKT in an S1P-dependent manner, while inhibition of JNK attenuates SK1-I-mediated cell death, 114 implying that targeting SPHK1 to induce cancer cells apoptosis requires cooperation with the JNK pathway. In gastric cancer cells, SPHK1 inhibits apoptosis by downregulation of Bim via stimulating Akt/FoxO3a signaling. 47 Through inhibiting SPHK1 by Ski (also SKI-II), SPHK1 protects multiple myeloma (MM) cells against apoptosis by inhibition of cancer-specific RTKs. 54 Interestingly, inhibition of SPHK1 also promotes the synthesis of a novel pro-apoptotic molecule diadenosine 5′,5′′′-P1, P3-triphosphate (Ap3A), which is associated with tumor suppressor of fragile histidine triad (FHIT), then triggering apoptosis. 110
Some non-coding RNA is associated with apoptosis, such as miRNA and LncRNA, and their mechanisms may be different. Overexpression of miR-515-5p in breast cancer cells could weaken SPHK1 activity, decreased cell proliferation, and elevated caspase-dependent apoptosis. 21 It has been shown that SPHK1-siRNA not only accelerates apoptosis but also restricts proliferation in ovarian cancer cells, 115 which could be a potential targeted therapy. 116 LncRNA HULC inhibits apoptosis by upregulating SPHK1 and its downstream PI3K/Akt pathway. 25 Downregulating lncRNA MEG3 inhibits apoptosis by upregulating TGF-β1 and its downstream SPHK1, 41 suggesting that the SPHK1-dependent inhibition of apoptosis in cancer cells could be a therapeutic target by different approaches, such as promoting hydrolysis, inhibiting translocation to plasmalemma, regulatory RNA, and chemical inhibition (Figure 3B).
In summary, SPHK1 serves as both a stimulant of autophagy and an inhibitor of apoptosis (Figure 3A/B/C). Autophagy protects cells from accumulating toxins, improper molecules, and damaged organelles and facilitates tissue development and cell differentiation. 117 Autophagy also supplies nutrition for cell metabolism by recycling the materials mentioned above. Under adverse conditions such as severe inflammation, hypoxia, or malnutrition, autophagy may become excessive and cause autophagic cell death. 108 SPHK1 may regulate autophagy and related signaling pathways in response to changes in the environment, leading to cell survival or death. Thus, targeting SPHK1-mediated autophagic cell death could be a promising therapeutic approach for cancer treatment.
Since SPHK1 also exerts anti-apoptosis effects, inhibitors targeting SPHK1 have been tested in various cancer models (Table 3). Furthermore, SPHK1-mediated autophagy may directly affect apoptotic signals (Figure 3C). It is widely believed that SPHK1-mediated autophagy and apoptosis are mutually exclusive. 118 For example, SPHK1-induced autophagy protects against apoptosis during nutrient starvation, while Cer-induced autophagy does not. 119 Autophagy usually precedes apoptosis and activates apoptosis via various mechanisms, but autophagy can also protect cells from apoptosis. Apoptosis and autophagy have distinct signaling pathways and cellular processes but also share some functions. 120,121 These 2 processes can occur simultaneously or independently, and the result depends on the environment and cellular response. 120,122 Altogether, either SPHK1-mediated autophagy or apoptosis can be targeted in cancer therapy to induce autophagic or apoptotic cell death (Figure 3C).
SPHK1 inducing tumor proliferation and survival
The proliferation of tumor cells is regulated by numerous cellular factors. 123 Cumulating studies have shown that SPHK1 promotes cell proliferation and tumor growth 18,88 (Figure 4).
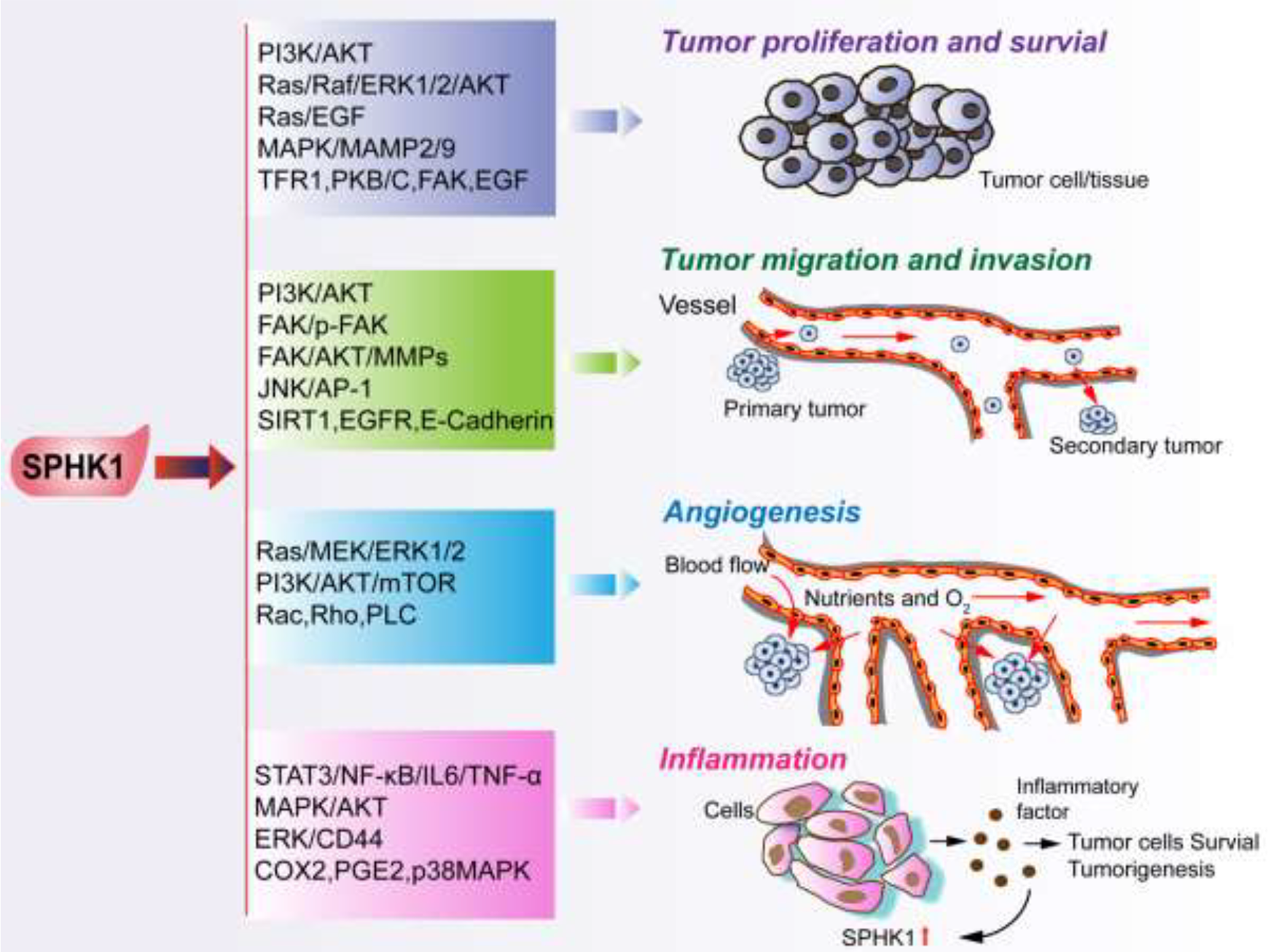
The mechanism of SPHK1 in proliferation, migration, invasion, angiogenesis, and inflammation.
SPHK1 is a positive regulator of cell proliferation and survival. Knocked-out of SPHK1 suppresses AKT signaling pathway and inhibits cell proliferation. 124 SPHK1 regulates tumorigenesis and tumor growth in early colon cancer, 125 glioma, 126 breast cancer, and chronic lymphocyte by modulating calcium signaling. 127,128,129 Overexpression of SPHK1 enhances triple-negative breast cancer proliferation via PI3K/AKT signaling, 130 whereas TNF-α regulates breast cancer cell proliferation by modulating Cer. 131 Elevated SPHK1 also augments colon cancer cell proliferation by regulating the MAPK/MMP-2/9 pathway. 71 Gene expression array has shown that SPHK1 regulates cell survival, proliferation, and tumor transformation by upregulation of transferrin receptor 1 (TFR1). 3 TFR1 is a novel target of SPHK1, which provides new insights into the regulatory mechanism of SPHK1-dependent tumorigenesis. Epoxyeicosatrienoic acid (EET) is a product of cytochrome P450 (CYP) peroxidase. Studies have shown that SPHK1 augments EET-stimulated endothelial cell proliferation, which requires S1PR1 and S1PR3. 132 Vascular endothelial growth factor (VEGF) can induce endothelial cell proliferation through its receptors. 133 Activated SPHK1 in breast cancer upregulates VEGF expression, and both VEGF and SPHK1 are independently regulated by the mammalian target of rapamycin C1 (mTORC1). 134 VEGF and follicle-stimulating hormone (FSH) could upregulate S1P synthesis through phosphorylation of SPHK1, accelerating cell proliferation. 135 Recent studies have revealed that inhibition of SPHK1 attenuates proliferation and survival in cancer cells by impairing PKC activity and cytokinesis. 62 Leptin is an essential adipokine that plays a key role in regulating energy balance, body weight, metabolism, and endocrine function. 136 Studies have shown that leptin can escalate the proliferation and activation of SPHK1 via ERK1/2 and Src family kinase (SFK) pathways. 137
MiRNAs such as miR-515-5p and miR-124 can inhibit apoptosis in tumor cells, and miRNAs can control the activity and expression of SPHK1 to suppress proliferation, which is closely related to WNT pathway. 21,31,138 In bladder cancer, miR-613 inhibits bladder cancer cells’ proliferation by targeting SPHK1 expression and function. 71,139 MiR-128 also suppresses the growth of thyroid carcinoma by downregulating SPHK1.56 Thus, SPHK1 is a target for miRNA with anti-cancer potential, 44 but further investigation of the miRNA-dependent regulation mechanism of SPHK1 is needed. Apart from miRNAs, lncRNAs with distinct functions significantly contribute to SPHK1 signaling as well. Particularly, lncRNAs HULC induces NSCLC proliferation and inhibits apoptosis by enhancing SPHK1 and PI3K/Akt signaling. 25 In contrast, suppression of lncRNA MEG3 accelerates colorectal adenocarcinoma cell proliferation and abolishes apoptosis by upregulating TGF-β1/ SPHK1 pathway. 41
Cancerous proliferation is often caused by cell cycle regulation abnormalities and genetic changes. 123,140 However, SPHK1 and SPHK2 activity are not required for tumor cell viability. 141 Altogether, it still needs to explore further the contribution of SPHK1 to tumor proliferation and cell survival. In summary, SPHK1 promotes cell proliferation and survival in cancer, whereas its inhibitors such as miRNA and chemical inhibitors suppress proliferation. These data provide a strategy to inhibit tumor proliferation by targeting SPHK1.
SPHK1 promoting tumor migration and invasion
SPHK1 has been implicated in tumor metastasis as well as invasiveness.38 SPHK1 is overexpressed in many tumor types (Table 2) and plays an important role in tumor migration and invasion. Previous studies have found that SPHK1 is overexpressed in NSCLC and enhances the migratory and invasive ability of NSCLC through the AKT pathway. 26 SPHK1 is also overexpressed in triple-negative breast cancer (TNBC). 23 SPHK1 knock-out attenuates migration and invasion of TNBC by controlling PI3K/AKT signaling pathway. 130 Tumorigenesis is not only triggered by intracellular factors but also closely related to the extracellular environment. 142 Hormones and growth factors can regulate SPHK1 and S1P production and facilitate the migration of endothelial cells. 143 Hepatocyte growth factor (HGF) could induce the phosphorylation of SPHK1 and drive the production of S1P, thereby promoting the migration of lung endothelial cells. 144 SPHK1/S1P regulates sirtuin 1 (SIRT1) expression through P38 MAPK, ERK, and AKT signals, and SIRT1 knock-out blocks the migration of human umbilical vein endothelial cells (HUVEC). 145 Death receptor 5 (DR5) is another SPHK1 functional partner. When DR5 is suppressed, SPHK1/S1P may be involved in TRAF2-mediated activation of JNK/AP-1 to stimulate tumor invasiveness. 146 SPHK1-dependent migration may also be associated with myristoylated alanine-rich C-kinase substrate (MARCKS)-related proteins. 147 These results show that SPHK1 promotes tumor migration and invasion in cooperation with other proteins. In response to irradiation, head and neck squamous cell carcinoma (HNSCC) migration may be associated with EGFR. 148 SPHK1 is also strongly linked to colon cancer cells (CRC), which advances CRC migration and invasion, and may be connected to E-cadherin and vimentin to induce EMT. 149 Similarly, SPHK1 can promote CRC migration and EMT by regulating the expression of p-FAK. 150 It was unveiled that SPHK1 facilitates colorectal cancer’s metastasis facilitates the metastasis of colorectal cancer by stimulating EMT via the FAK/AKT/MMPs axis. 42
The production of S1P and its receptor S1PR are also associated with migration and invasion regulated by SPHK1. When S1P is produced in a large amount, it also provokes migration of oral squamous cell carcinoma (OSCC), at least through targeting S1PR2. 151 SPHK1-mediated renal clear cell carcinoma (ccRCC) invasion occurs by S1PR2-dependent FAK phosphorylation and a FAK-independent mechanism through S1PR1/3.46 And HNSCC invasion possibly associates with S1PR1. 152 By the cancer genome atlas (TCGA) RNA database analysis, SPHK1 stimulates invasion in von Hippel-Lindau (VHL) mutant ccRCC via S1PR2-dependent FAK phosphorylation and FAK-dependent S1PR1/3 mechanism. 46
Viruses are also implicated in SPHK1-mediated tumor invasion. 153 Hepatitis B virus (HBV) contributes to tumor growth by upregulating AP2α and SPHK1, 154 and SPHK1 can trigger the proliferation, growth, and migration of HCC through S1P/endothelial differentiation G-protein coupled receptor 1 (EDG1) and nuclear factor Kappa B subunit 1 (NF-κB) pathways. 155 Moreover, Epstein-Barr virus (EBV) activates AKT via the SPHK1/S1P pathway to promote the migration of undifferentiated nasopharyngeal carcinoma (NPC). 156 These findings suggest that pathogenic microorganisms should be considered as a novel regulator of SPHK1 in tumorigenesis.
SPHK1-related miRNAs also are associated with tumor migration and invasion. Overexpression of SPHK1 upregulates miRNAs that play different roles in tumorigenesis. In particular, the miR-144-3p/fibronectin 1 (FN1) pathway may be involved in the pro-invasive role of SPHK1 in thyroid papillary cancer cells (PTC). 157 Of interest, some miRNAs show distinct effects. For example, miR-613 protect against migration by targeting SPHK1 in bladder cancer 139 and PTC, 158 while miRNA-124 targets SPHK1 and down-regulates SPHK1 expression to arrest tumor proliferation and migration. 138
SPHK1 is currently explored as a therapeutic target to restrain tumor growth and migration. Dimethylsphingosine (DMS, SPHK inhibitor) can govern tumor invasiveness through NF-κB, alleviating tumor permeation. 159 CB5468139 also inhibits migration by targeting SPHK1 (Table 3). These results provide an anti-metastasis therapeutic strategy by targeting SPHK1. In conclusion, SPHK1 promotes tumor migration and invasion with viral infection as an important factor, which calls for further study into the detailed regulatory mechanism.
SPHK1 prompting tumor angiogenesis
To ensure tumor growth, the tumor tissue will establish a vascular system, in which the tumor or other cells secrete pro-angiogenic factors and facilitate angiogenesis. 160 SPHK1 promotes tumor angiogenesis by several mechanisms (Figure 4).
SPHK1 and its metabolite S1P significantly contribute to tumor angiogenesis and survival. 71 In epithelial ovarian cancer, inhibition of SPHK1 or S1PR1/3 attenuates angiogenic potential and angiogenic factor secretion. 161 When S1P is added, angiogenic potential and angiogenic factor secretion can be restored, suggesting that the SPHK1/S1P/S1PR1/3 pathway plays an essential role in angiogenesis. 30 The identical mechanism was also characterized in clear cell renal cell carcinoma (ccRCC). 46 Angiogenesis is associated with cancer metastasis and invasion, while the molecular mechanism is not yet clear. 162 Enhancing the activity of SPHK1 triggers angiogenesis and facilitates invasion and metastasis of cervical cancer. 163 Increased SPHK1 leads to upgraded S1P production, resulting in angiogenesis and metastasis in triple-negative breast cancer 23,128 and undermine breast cancer migration and survival. 164 S1P targets corresponding receptors in hematologic malignancies, activates Ras/MEK/ERK1/2, PI3K/AKT/mTOR, Rac, Rho, and PLC to govern angiogenesis. 165 Besides, SPHK1 promotes angiogenesis, metastasis, and invasiveness in head and neck squamous cell carcinoma (HNSCC). 166
Besides, non-coding RNAs, such as miRNAs and lncRNAs, are also involved in SPHK1-mediated angiogenesis. As an example, miR-506 can suppress the expression of SPHK1 in hepatocellular carcinoma HepG2 cells through binding to the 3′-UTR of SPHK1-mRNA and reduce S1P production to impair angiogenesis. 37 Another lncRNA has also been demonstrated to promote tumor angiogenesis in liver cancer by upregulating SPHK1. 167
In summary, SPHK1 prompts tumor angiogenesis, which provides new capillaries to deliver oxygen and nutrients to the tumor (Figure 4) and facilitates metastasis.
The relationship between SPHK1 and inflammation
SPHK1 and S1P are closely associated with inflammation and inflammatory factors. 168,169 Especially, inflammatory factors such as cytokines trigger inflammation and tumorigenesis by activating SPHK1. 88 Therefore, SPHK1 could be viewed as a mediator of inflammation in the tumor microenvironment.
The SPHK1/S1P pathway is involved in inflammatory responses to cytokines, such as TNF-α and IL-1. 18 SPHK1 can also be activated by inflammatory signaling molecules such as IL-1b, IFN-g, and IgE. 170 However, SPHK1 knock-out mice lack systemic inflammatory response. 171 TNF-α activates SPHK1 via a TRAF2-dependent mechanism and affects cell inflammation and survival through AKT and NF-κB signaling. 18 In a mouse model, SPHK1 and TRAF2 regulate TNF signaling, and inhibition of NF-κB and TNF signaling may be beneficial to certain diseases. 113 Prior research suggests that SPHK1 functions through the p38MAPK pathway and was not required to activate the canonical NF-κB pathway. 172 Therefore, the molecular mechanism of SPHK1 in inflammation and tumors needs further investigation. Of course, SPHK1 can promote the release of pro-inflammatory cytokines such as IL-6 in turn. 18,173 It is reported that SPHK1 mediated inflammation and colon cancer by modulating various molecules, including cyclooxygenase 2 (COX2), prostaglandin E2 (PGE2), NF-κB, IL-6, and signal transducer and activator of transcription 3 (STAT3). 174 For COX2, it suppresses the anti-tumor response by CTL-, Th1-, and NK cell-mediated type-1 immunity. 175 But whether the role of COX2 in inflammation is caused by SPHK1 phosphorylation or acetylation is not clear since SPHK1 acetylates COX2 in an acetyl-CoA dependent manner. 66 STAT3 is a key transcription factor of inflammation and cancer. 166 SPHK1/S1P is implicated in the regulation of STAT3 and the development of intestinal inflammation and relevant cancers, 176 and it upregulates CD44 in human colon cancer cells through ERK signaling. 43 Targeting S1P signaling and metabolism might be a new strategy for treating inflammatory bowel disease (IBD), which may lower colon cancer risk. 177 In the mouse intestinal cancer model, the upregulation of SPHK1 and S1P production is beneficial to developing inflammation and colon cancer via the signaling network of S1P/S1PR1/STAT3/NF-κB/IL-6/TNF-α. 178 SPHK1 is also associated with the development and progression of inflammatory gastrointestinal (GI) diseases, leading to GI cancer through immunologic mechanisms, 179 suggesting SPHK1 as a pivotal regulator of inflammation-induced tumorigenesis (Figure 4).
SPHK1 is originally recognized as a pro-inflammatory enzyme capable of activating TNF-α and NF-κB, 180,181 but later studies have identified that in the mouse bone marrow macrophage (BMDM) model, SPHK1 is not essential for inflammatory response. 182 Aquaporin 4 (AQP4) is functionally associated with SPHK1 directly or indirectly. AQP4 deficiency was associated with the SPHK1/MAPK/AKT pathway and could alleviate the release of pro-inflammatory cytokines from astrocytes. 183 Targeting the SPHK1/S1P/S1PR1 pathway, which influences the progression of inflammation and breast cancer, is dependent on the activation of STAT3 and upregulation of IL-6. 184
In conclusion, SPHK1 strongly correlates with inflammation. In general, pro-inflammatory molecules activate the SPHK1/S1P signaling pathway that upregulates STAT3, NF-κB, IL, TNF-α, MAPK, AKT, ERK, COX2, etc. which in turn promote cytokines production and release. Last, the amplification loop promotes prolonged and excessive inflammation that facilitates tumorigenesis. Here we speculate positive feedback between SPHK1 and inflammation. Inflammatory signals activate SPHK1 that subsequently promotes the synthesis and release of inflammatory molecules to the tumor microenvironment, which could promote tumorigenesis and cancer cell survival (Figure 4).
The Recent Development of Potential SPHK1 Inhibitors
Due to the association of SPHK1 with various processes of tumorigenesis, inhibition of SPHK1 is an attractive approach to limit tumor growth and metastasis. A wide variety of potent agents targeting SPHK1 have been developed, including SK1-I, SKI-I∼V, DMS, FTY720, Safingol, SKI-178, SK-F, B-5354c, F-12509a, CB5468139, SLP7111228, Compound 23/51/82, and PF-543 (Table 3, Figure 5). The summary of these drug candidates is described below.

The structure of Sph, S1P, and SPHK1 inhibitors. A, The structure of Sph, S1P. Both Sph and S1P are sphingolipids. The former is the substrate of the SPHK1 enzyme, and the latter is the production of SPHK1. B, The structure of dual SPHK1/SPHK2 inhibitors. C, The structure of SPHK1-selective inhibitors.
Dimethylsphingosine (DMS), the first direct SPHK inhibitor, suppresses cancer cell growth, and induces apoptosis while also potently blocks PKC signaling. 185 Meanwhile, DMS has been shown to inhibit both SPHK2 and ceramide kinase (CERK), making it unable to decipher the role of SPHK1. 11 (2 R, 3 S, 4E)-N-methyl-5-(4-pentylphenyl)-2-aminopent-4-ene-1, 3-diol (SK1-I), an Sph-competitive SPHK1 inhibitor, has been widely implemented in vivo to elucidate the role of SPHK1 in cancer. 186 SK1-I decreases serum S1P levels, stimulates cancer cell apoptosis, and prevents lymph node and lung metastasis in a murine breast cancer model. 187 SK1-I is regarded as a selective SPHK1 inhibitor that does not suppress SPHK2, 188 but it is also reported to restrain SPHK2 with a similar affinity. 189 Although SKI-I (N-[(2-hydroxy-1-naphthyl)methylene]-3(2naphthyl)-1Hpyrazole-5-carbohydrazide) is also an Sph-competitive inhibitor of SPHK1, it targets SPHK2 and cross-reacts with ERK2, PKC, and PI3 K. 186 Similar to SKI-I, 2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole (SKI-II, also SKi) was revealed to attenuate SPHK1 signaling by triggering the lysosomal degradation of SPHK1 in various cell types rather than direct inhibition. 190 However, SKI-II targets not only SPHK1 but also impairs SPHK2 activation. 186 Certainly, it was more effective to suppress SPHK1 than SPHK2 based on their Ki (Ki -SPHK1 = 16, Ki-SPHK2 = 8). Moreover, SKI-II decreases cellular S1P levels and increases cellular Cer and Sph, leading to apoptosis and suppression of proliferation and migration. 191 Another pharmacologic inhibitor, SKI-III∼V, an analog of SKI-I/II, has been unveiled by a series of modifications of Sph. 192
Fingolimod (FTY720), identified by Billich et al. in 2003, 193 is an FDA-approved immunomodulating drug for multiple sclerosis by antagonizing S1PR1. 194,195 FTY720 inhibits S1P generation by competition with Sph as a substrate for both SPHK1 and SPHK2, 69 with a lower efficiency with SPHK1 than SPHK2. 196 FTY720 also promotes the degradation of SPHK1 via ubiquitination and proteasomal pathway in cell lines. 18,196 In vitro and in vivo studies demonstrate the anti-growth and pro-apoptosis ability of FTY720 in various normal and cancer cells. 196 In a model of human ALL xenografts in mice, FTY720 remarkably reduces the incidence of leukemia, which may be a potential treatment for Ph+ ALL but not Ph- B-ALL. 197 L-threo-dihydrosphingosine (Safingol, also DHS) showed potent SPHK-inhibiting properties 198 and was a promising anti-cancer agent, but it lacks specificity, with SPHK1, SPHK2, and PKC as targets. 199
More novel and potent inhibitors of SPHK1 are in the pipeline (Figure 5). It is reported that SKI-178 suppresses the functions of SPHK1 in tumorigenesis and cancer progression in vitro and in vivo. 198 It is SPHK1-selective and has low toxicity (Table 3). SK-F is another selective inhibitor of SPHK1, which potently decreases cancer cell viability in vitro and sensitized mouse breast tumors to docetaxel (DTX) in vivo without significant whole-body toxicity. 198 F-12509a, a new sphingosine kinase inhibitor, led to ceramide accumulation, reduction in S1P content, and apoptosis. 200 F-12509a functions by mimicking the conformation of Sph to bind to the active site of SPHK1. In contrast, B-5354c, a non-competitive inhibitor for Sph, maybe interact with domains distinct from the Sph associating sites to regulate SPHK1 activity. 185 Of note, B-5354c and F-12509a are isolated from a marine bacterium and discomycete Trichopezizella barbata, respectively. 201
The ATP-competitive SPHK1 selective inhibitor, CB5468139, was identified from a small molecule library. 191 Unfortunately, CB5468139 blocks various protein kinases such as CDC like kinase 1 (CLK1), Src family tyrosine kinase (FYN), Met (tyrosine kinase), MST2 (histone acetyltransferase), PIM2 (serine/threonine kinase), SYK (spleen associated tyrosine kinase) and tyrosine kinase non-receptor 2 (TNK2) (with IC50 values<2 μM). 186 Thus, the phenotypes observed after treatment may be due to off-target effects. Compound 23 was identified from a series of structure-based SPHK1 and SPHK2 inhibitors. It has a lower IC50 of 20 nM to SPHK1 than other compounds and makes key hydrogen bonding interactions with D81 and D178 of SPHK1. 202 Similar with compound 23, compound 82 has the same IC50 to SPHK1 and reduces plasma S1P levels by approximately 3.5 fold. 202 Unfortunately, its effects on endogenous lipids or protein kinases have not been evaluated. 186 SLP7111228 (compound 36a, Ki = 48 nM) is a potent and selective SPHK1 inhibitor, which decreases S1P levels in U937 cells. Administration of SLP7111228 in rats also reduces blood S1P levels. 203 Compound 51 is an SPHK1-selective inhibitor with improved aqueous solubility and ADME properties while maintain or enhance enzyme potency. 192 Notably, a novel cell-permeant inhibitor of SPHK1, PF-543, is the most potent inhibitor of SPHK1 with 130-fold selectivity over SPHK2. 204,205 PF-543 has been co-crystallized with SPHK1, which gives structural insights into the Sph pocket and suggests that Phe302 in the SPHK1b isoform is not conserved in SPHK2, confers selectivity for SPHK1. 206 The development of PF-543 has shown that even if S1P is strongly reduced, there is no effect on cancer cell proliferation.
Altogether, most SPHK inhibitors lack specificity to SPHK1 (Table 3, Figure 5). Recently, novel molecules such as SKI-178, B-5335c, PF-543, and compound 51/82 are designed to target SPHK1 (Table 3) selectively. However, these second-generation inhibitors would need validation in tumor growth and metastasis models first.
Currently, there are no FDA-approved drugs that target SPHK1. Most SPHK1 inhibitors remain at the pre-clinical stage, except Safingol (Table 3). Safingol has concluded Phase I trial with non-toxic and a maximally tolerated dose. 185 Of note, S1PR1 antagonist FTY720 was approved by the FDA in 2010 for treating multiple sclerosis. 194
Conclusion
In summary, SPHK1 is a crucial sphingolipid metabolic kinase with versatile functions, especially in tumor biology. SPHK1 regulates tumorigenesis and cancer progression by modulating various cellular processes, including apoptosis, autophagy, proliferation, migration, invasion, angiogenesis, and inflammation, suggesting SPHK1 as a promising target for cancer therapy. Further investigation into the detailed oncogenic mechanism of SPHK1 and the development of potent SPHK1 inhibitors with improved specificity may offer a novel direction in cancer therapy.
Footnotes
Author Contributions
Xianwang Wang, PhD, Yong Sun, MM and Xiaochun Peng, PhD are authors contributed equally to this work. XW W, XC P, YY L, and SY conceived the study. YS performed the literature search and data analysis. YS, SM Abbas, YY, JZ, MW C, YC, HY C, HY Y, and GL W produced the figures and tables. XW W, PH, YY L, and SY wrote the manuscript. All authors read and approved the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (31700736, 31501116), China Scholarship Council (201908420102), Hubei Province Scientific and Technological Research Project (D20201306), Hubei Medical Youth Tip-Top Talent, Leading Talent Program of Yangtze Talent Project and the College Students Innovative Entrepreneurial Training Program in Yangtze University (2018184, 2019372, Yz2020334).