
Abstract
Fas-associated protein with death domain (FADD) was originally reported as a proapoptotic adaptor molecule that mediates receptor-induced apoptosis. Recent studies have revealed a potential role of FADD in NF-κB activation, embryogenesis, and cell cycle regulation and proliferation. Overexpression of FADD and its phosphorylation have been associated with the transformed phenotype in many cancers and is therefore a potential target for therapeutic intervention. In an effort to delineate signaling events that lead to FADD phosphorylation and to identify novel compounds that impinge on this pathway, the authors developed a cell-based reporter for FADD kinase activity. The reporter assay, optimized for a high-throughput screen (HTS), measures bioluminescence in response to modulation of FADD kinase activity in live cells. In addition, the potential use of the reporter cell line in the rapid evaluation of pharmacologic properties of HTS hits in mouse models has been demonstrated.
Introduction
FADD (F
Phosphorylation of FADD at serine 194 has been shown to be mediated by casein kinase Iα (CK1α) 9 by FIST (FADD interacting serine threonine kinase), 10 as well as by PKC-zeta. 11 CK1α and FIST are the FADD kinases commonly identified in cancer cells. FIST activity can be downregulated in response to JNK inhibition, 10 resulting in decreased FADD phosphorylation. In addition, CK1α inhibitors (CKI-7) also inhibit FADD phosphorylation. However, the regulation and role of FADD kinases in cancer is not well understood. To identify lead compounds that modulate FADD phosphorylation as potential therapeutic agents and to better define molecular events that lead to FADD phosphorylation, we describe here the development of a molecular imaging-based reporter for FADD kinase activity (FADD kinase reporter, FKR) and the optimization of the cell-based assay for high-throughput screening (HTS).
Experimental Procedures
Plasmid construction
FKR was generated in the mammalian expression vector pEF. N-terminal firefly luciferase gene (NLuc) was amplified by PCR, 12 using primers that generated a product comprising a SalI restriction site followed by a Kozak consensus sequence, with a NotI restriction site at the 3′ end. FHA2 was amplified from the Rad53p FHA2 domain with a sense primer containing a NotI site and a reverse primer containing an XbaI site. C-terminal firefly luciferase gene (C-Luc) was amplified using primers that produce a 5′ XbaI followed by the FADD kinase substrate sequence, 142-208 amino acids harboring serine 194, flanked by a linker (GGSGG) at each side, with a 3′ EcoRI restriction site after the terminating codon. FKR mutant was constructed by changing the phosphorylation site, serine 194, to alanine using the QuickChange kit (Stratagene, La Jolla, CA). All plasmids were verified by automatic DNA sequencing.
Cell culture and transfection
The human head and neck squamous carcinoma (UMSCC-1) cells were provided by Thomas Carey, Head and Neck Oncology Program, University of Michigan. Human lung epithelial carcinoma (A549) cells were purchased from the American Type Culture Collection. UMSCC1 and A549 (lung cancer) cells were grown in RPMI-1640 (Invitrogen, Carlsbad, CA) and Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen), respectively. Complete medium was supplemented with 10% heat-inactivated fetal bovine serum (FBS; GIBCO, Carlsbad, CA) and 100 U/mL penicillin/streptomycin. Cell cultures were maintained in a humi-dified incubator at 37°C and 5% CO2.
To construct stable cell lines, the FKR plasmids (wild type and mutant) were stably transfected into UMSCC1 and A549 cells using Fugene (Roche Diagnostics, Indianapolis, IN), and stable clones were selected with 500 µg/mL G418 (Invitrogen). Resulting clones were isolated and cultured for further analysis by Western blot for determination of expression levels of the recombinant protein.
Antibodies and reagents
Rabbit polyclonal antibodies to phospho-FADD and c-Jun were purchased from Cell Signaling Technology (Danvers, MA). Goat polyclonal antibodies to CK1α and firefly luciferase were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Chemicon (Millipore, Billerica, MA), respectively. Mouse monoclonal antibodies to FADD were purchased from BD Pharmingen (San Diego, CA). Actin antibody was purchased from Sigma Aldrich (St. Louis, MO).
JNK Inhibitor (SP600125) was obtained from Calbiochem (EMD Chemicals, San Diego, CA) and CKI-7 from Toronto Chemical Research (New York, Ontario, Canada). Luciferin was obtained from Biosynth (Naperville, IL).
Small interfering RNA transfection analysis
Small interfering RNA (siRNA) sequences for sense strand CK1α and FIST/HIPK3 were 5′-CCAGGCAUCCCCAGUUG CUTT-3′, 5′-AAU ACU UAC GAA GUC CUU CAU-3′, respectively, and were synthesized from Invitrogen. FADD siGENOME Smart Pool and nonsilencing siRNA (NSS) were synthesized by Dharmacon Research (Lafayette, CO). siRNA transfection was carried out using Oligofectamine (Invitrogen) according to the manufacturer’s instructions. Cells were analyzed 48 to 72 h later for bioluminescence imaging followed by Western blot.
Western blotting and immunoprecipitation
UMSCC1 FKR cells in culture dishes were collected and centrifuged at 1800 g for 5 min at 4°C. Cell pellets were washed twice with cold phosphate-buffered saline (PBS) and then lysed with a buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 50 mM NaF, and 1 mM Na3VO4 and supplemented with complete protease inhibitors mixture (Roche Diagnostics). Cells in lysis buffer were rocked at 4°C for 30 min. The lysates were then cleared by centrifugation. The supernatants collected were estimated for protein content by a detergent-compatible protein assay kit from Bio-Rad (Hercules, CA). Lysates with equal amounts of protein were separated by Laemmli sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the levels of protein expression were detected by Western blot analysis with anti-FADD (1:2000), p-FADD (1:1000), CK1α (1:200), FIST (1:500), luciferase (1:5000), and actin (1:1000). Specific signals were visualized by using the Enhanced Chemiluminescence (ECL) Western Blotting System (GE Healthcare, Piscataway, NJ).
For immunoprecipitation, cell extracts (2000 µg) were incubated with the luciferase antibody for 1 h. Immune complexes were captured using protein G–Sepharose (GE Healthcare) and washed 3 times using RIPA buffer. The resulting pellet was boiled for 5 min in sample buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue, 0.125M Tris-HCL, pH 6.8) and resolved by SDS-PAGE.
96-well assay
A549 FKR cells were seeded in 96-well plates, using a Titertek Multidrop Microplate Dispenser (Thermo Fisher Scientific, Waltham, MA) and incubated 24 h prior to compound addition. Compound stocks were prepared in DMSO and diluted 1:100 in PBS. Then, 10 µL of the intermediate stock was added to each assay plate to achieve final concentrations from 3 nM to 50 µM using the Beckman Biomek NXP Laboratory Automation Workstation (Beckman Coulter, Fullerton, CA). The cells were incubated with test compounds at 37°C, 5% CO2 for 6 h. The final DMSO concentration was 0.1%.
Variability tests were conducted on 3 types of signals. Maximum and minimum signals were obtained by treating plates with 30 µM JNK inhibitor and DMSO, respectively. To estimate signal variability at points between the maximum and minimum signals, JNK inhibitor was used at a concentration of 15 µM. Results were analyzed by plotting the signal from each well against the well number in which the wells were ordered by row and then by column.
In vivo studies
A549 FKR (1.0 × 106) cells were transplanted into the flanks of nu/nu CD-1 nude mice (Charles River Laboratories, Wilmington, MA). When tumors reached approximately 100 mm3 in volume, the mice were randomized into 3 groups, and treatment was initiated. A CK1α inhibitor in DMSO was given via intraperitoneal injection at doses of 0.2 and 0.5 mg/kg. DMSO was used as control.
Bioluminescence imaging
Live-cell luminescent imaging was achieved by adding D-luciferin (200 µg/mL final concentration) to the assay medium following compound incubation. Photon counts were acquired 5 to 10 min after 37°C incubation with D-luciferin using the IVIS imaging system (Caliper Life Sciences, Hopkinton, MA). An exposure time of 1 min was used for acquisition. Living Image 3.0 software was used for data analysis (Caliper Life Sciences). For in vivo bioluminescence, mice were anesthetized using a 2% isofluorane/air mixture and injected with a single dose of 150 mg/kg D-luciferin in PBS intraperitoneally. Image acquisition was initiated 5 min following injection of luciferin. Consecutive bioluminescence images were acquired before treatment and then every 3 h for 9 h.
Data analysis
Data were collected from at least 2 independent experiments with 3 or more replicates per experiment. Graph Pad Prism v.5 nonlinear regression analysis (GraphPad Software, San Diego, CA) was used to generate the 50% inhibition concentration (IC50) values. Fold induction was calculated as signal-to-noise ratios. The Z factor was calculated as previously described. 13
Results
Development and validation of bioluminescent FKR
Rather than study each of the known FADD kinases and their regulation in normal versus tumor cells individually, we chose to develop a pan reporter for FADD kinase activity (FKR) that would noninvasively report on changes in FADD kinase activity in living cells. This reporter was based on a platform we recently described
14
for imaging of Akt, except that the target peptide was substituted with residues 142-208 of FADD, which encompasses Ser194, the primary site for phosphorylation in FADD (
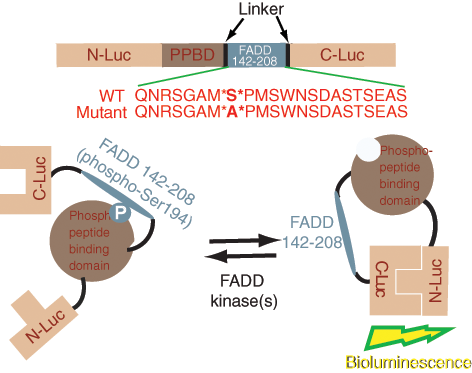
Construction of the FADD kinase reporter (FKR). The model shows a diagrammatic representation of the domain structure of FKR and the sequence of the C-terminal domain that harbors serine 194, the target for FADD kinases. N-Luc and C-Luc are the N- and C-terminal domains of firefly luciferase that are fused to the FHA2 appropriate ends of the reporter. Two versions of the FKR were developed: the FKR (WT), which contains the wild-type FADD peptide sequence, and the FKR (Mutant), which contains a serine 194 substitution to alanine at the primary phosphorylation site. Phosphorylation of the FADD peptide at serine 194 results in interaction with FHA2 phosphopeptide binding domain (PPBD) causing steric constraints on C-Luc and N-Luc. Inhibition of FADD kinase results in decreased binding of phosphopeptide and PPBD, enabling N-Luc and C-Luc interaction to restore enzymatic activity.
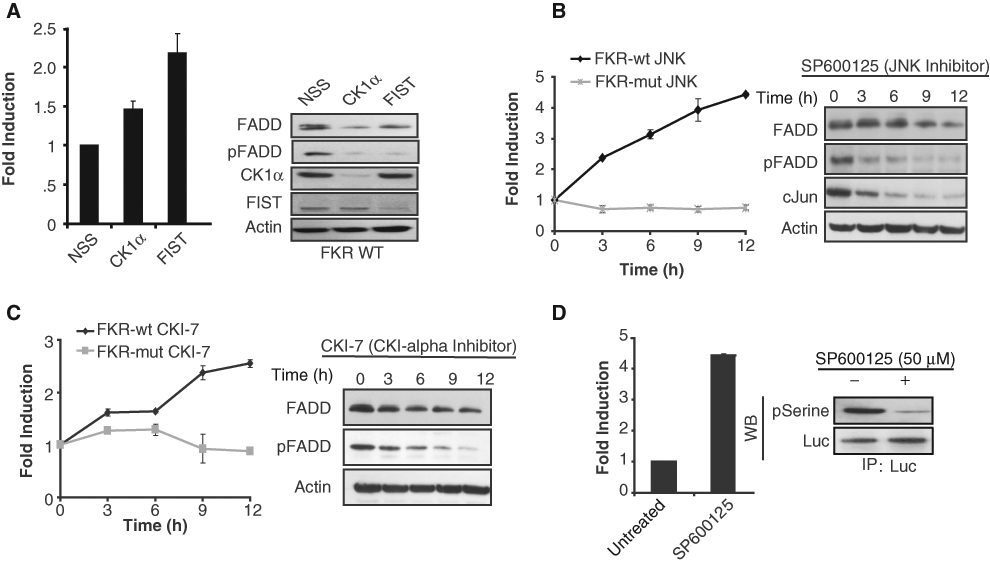
Validation of the FADD kinase reporter (FKR). (A) UMSCC1 FKR stable cells were transfected with siRNA targeting CK1α, FIST/HIPK3, or nonsilencing siRNA (NSS). Bioluminescence was measured after 48 h of transfection. Cell lysates were prepared and analyzed by Western blotting using antibodies specific for FADD, phospho-FADD, CK1α, FIST/HIPK3, or actin as a loading control. (
HTS optimization and validation
A549 FKR bioluminescence activity and background response was compared at seeding densities from 5000 to 12,000 cells per well in 96-well plates. Bioluminescence intensity was found to increase proportionally to cell number, indicating that signal intensity correlated directly with cell number per well (
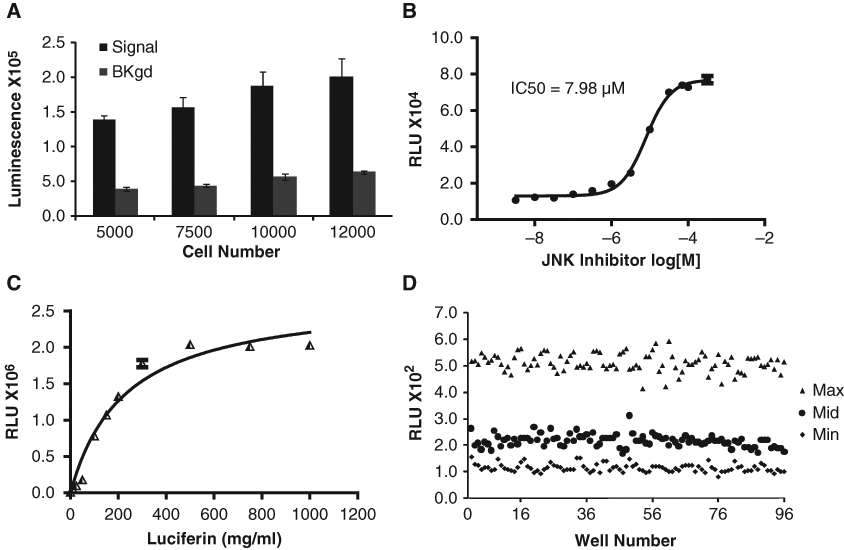
High-throughput screening (HTS) optimization. (
A validation screen, results of which are shown in
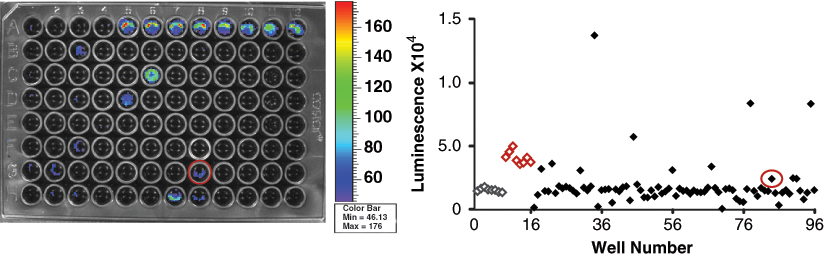
A549 FADD kinase reporter (FKR) validation screen. The 80-compound screen was conducted under optimized assay conditions. Positive control JNK inhibitor (SP600125) was placed in wells A5-A12 and negative control, DMSO in wells A1-A4 and H9-H12 (controls identified on scatter plot by red and gray for positive and negative, respectively). Control compound JNK inhibitor, located in well G8 (circled), was part of the compound set and demonstrated bioluminescence activity in the validation screen, thereby demonstrating assay utility in identifying modulators of FADD kinase activity.
A549 FKR in vivo imaging
To demonstrate the utility of the reporter beyond HTS, A549 FKR cells were implanted subcutaneously as a xenograft model to assess the in vivo bioavailability and efficacy of FADD kinase inhibitors identified by the assay. When the A549 FKR tumors reached an approximate volume of 100 mm3, mice were treated with a CK1α inhibitor or with vehicle control. Bioluminescence activity was monitored over time. A time- dependent increase in bioluminescence was observed in mice treated with 0.5 mg/kg, whereas animals treated with 0.2 mg/kg did not show an increase over the 9-hour measurement period (
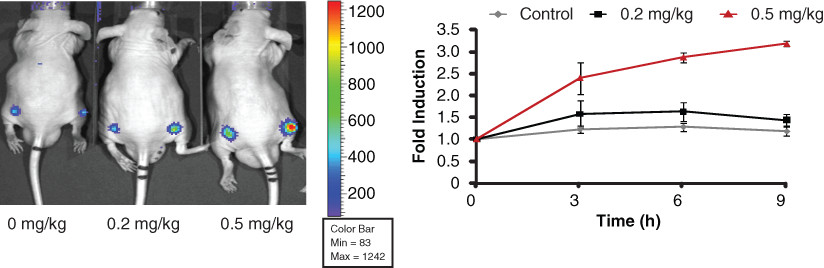
Time and dose imaging of FADD kinase reporter (FKR) activity. Representative image of CD-1 nude mice transplanted with A549 FKR cells and treated with 0.2 mg/kg and 0.5 mg/kg CK1α inhibitor. DMSO served as the control. Mice were imaged at 0, 3, 6, and 9 h following compound injection. Graph represents induction of bioluminescence at each time point. Fold induction was calculated for each time point as the ratio of bioluminescence activity following treatment and the basal bioluminescence prior to treatment. Data show a time- and dose-dependent increase in bioluminescence in vivo.
Discussion
In an attempt to define the molecular basis of high phospho-FADD levels in tumor versus normal tissue (in HNSCC and lung cancer), we developed FKR, a reporter wherein FADD kinase activity, irrespective of the kinase involved, could be quantitatively and noninvasively monitored in living cells. We found that inhibition of FADD kinase activity through the use of siRNA or by small molecule resulted in a decrease in levels of phospho-FADD and a corresponding activation of the reporter’s bioluminescence activity in a dose- and time-dependent manner. The design of the reporter wherein inhibition of kinase activity would result in activation of the reporter (a positive signal) provides numerous advantages. First, the signal to noise is improved, and second, false positives are minimized (cytotoxic compounds would appear as false positives in an assay wherein inhibition of a kinase would result in loss of reporter activity). In contrast, in cells expressing FKR-mut, wherein a serine to alanine substitution is introduced (corresponding to Ser194 of FADD), inhibition of FADD kinase activity does not result in a significant change in bioluminescence, thereby demonstrating the specificity of the reporter.
The luciferase complementation reporter system described here possesses 1/100th the activity of wild-type luciferase in the absence of complementation and as much as 1/10th the activity of wild-type luciferase activity in the presence of complementation. 16 Since the mutant reporter lacks the target phosphorylation site (serine mutated to alanine), it is constitutively active and therefore possesses a level of luciferase activity that is comparable to FKR in response to a FADD kinase inhibitor. In this regard, the mutant reporter possesses 1/10th the activity of wild-type luciferase. The presence of compounds in the library that stabilize the half-life of luciferase and/or enhance expression of the reporter by upregulating its transcription or translation would appear as positive hits in the FKR wild-type screen. Since these compounds exert their FADD kinase- independent activities equally on the mutant and the wild-type reporter, a secondary screen would identify them as false positives. Likewise, promiscuous or chemiluminescent compounds would also appear as positive hits in both screens. This makes the mutant reporter a useful tool in the identification of false positives from a high-throughput screen.
Further validation of the reporter was provided by studies demonstrating inhibition of JNK resulted in a 4-fold increase in bioluminescence and a corresponding decrease in serine phosphorylation of FKR, demonstrating that the reporter itself was a substrate for FADD kinases. Recent studies in our lab have demonstrated that phosphorylation of FADD results in its stabilization, whereas inhibition of FADD phosphorylation results in its ubiquitin-dependent degradation. Our studies using MG132, a proteosome inhibitor, have confirmed that a decrease in total FADD levels in response to FADD dephosphorylation (i.e., in the presence of a FADD kinase inhibitor) can be reversed by inhibiting proteosomal degradation (Suppl. Fig. S2).
The FKR allows for noninvasive monitoring of FADD phosphorylation in live cells quantitatively and dynamically in real time. There are numerous advantages to screening compounds in a live-cell assay as compared to a purified biochemical assay. These include the ability to evaluate drug-target interactions in a physiologically relevant context. For example, compounds unable to diffuse to the target site or are cytotoxic go undetected, thereby leading to their immediate elimination from further investigation. In addition, the bioluminescence readout can be used quantitatively to determine optimal time points to measure the response of interest. Although inherent toxicity is not detected in biochemical assays, live-cell assays enable the measurement of short- and long-term responses, which helps rule out off-target effects such as toxicity to cells. Biochemical assays, however, are advantageous for HTS due to parameters such as high sensitivity and the ability to define screening conditions.
Our data demonstrate that the A549 FKR signal-to- background ratio is independent of cell density within a given range, as evidenced by the proportional increase in signal and background with increasing cell density. When optimizing an assay to a high-throughput format, it is advantageous to incorporate flexibility into the screening protocol. Parameters including poor cell growth and differential seeding densities can affect assay results and reproducibility. However, our data demonstrate consistency in signal-to-background ratio independent of cell density and therefore allow for flexibility in cell plating conditions when conducting HTS. Treatment of cells with SP600125 (JNK inhibitor) results in a dose-dependent increase in bioluminescence, corresponding to a decrease in FADD kinase activity. The pharmacology of the assay can be used to measure day-to-day assay variability and to rank order compound hits according to potency. These optimization data were derived using the 96-well assay format; however, optimization for the 384-well format has been conducted and can be found online as Supplementary Figure S1.
The efficacy of FKR to selectively identify inhibitors of FADD kinase activity was demonstrated by the validation screen. The screen resulted in the positive identification of known FADD kinase modulators, that is, inhibitors of the mitogen-activated protein kinase (MAPK) pathway, previously shown to inhibit FADD phosphorylation in acute lymphocytic leukemia cells.
17
Our positive control JNK inhibitor (SP600125), a known FADD kinase inhibitor, was also part of the compound set and demonstrated activity in the assay (
Our experience has identified 2 major sources of false positives against FKR. The first source includes compounds with inherent luminescence, and the second source includes compounds that modulate signaling events upstream of the target. The former can be eliminated with the FKR-mut reporter. Resolution of the latter (i.e., how to identify compounds specific to the inhibition of one of the known FADD kinases as opposed to upstream events) would require a purified system. However, the ability of the screen to identify upstream signaling events could be beneficial in identifying signaling hubs that impinge on the target kinase.
Identifying compound hits from a high-throughput screen with desirable pharmacological properties requires tedious pharmacokinetic/pharmacodynamic and ADME/Tox studies and is both time-consuming and labor intensive. The in vivo results presented here allow for the identification of compounds with desirable pharmacologic properties by rapidly evaluating drug target interactions in vivo, thereby providing preliminary insight as to whether the drug is a potential therapeutic agent. The FKR cell-based assay platform will provide rapid identification of lead compounds from HTS, as well as providing in vivo validation of active compounds identified by HTS by rapidly validating drug-target interaction and dose and schedule optimization.
Footnotes
This work was supported by the US National Institutes of Health research grants R01CA129623 (AR), R21CA131859 (AR), U24CA083099 (BDR), and P50CA093990 (BDR) and by a developmental project to MSB by the National Institutes of Health through the University of Michigan’s Head and Neck SPORE Grant (5 P50 CADE97248).