
Abstract
In the cyanobacterial circadian clock, a core oscillator comprising the proteins KaiA, KaiB, and KaiC keeps time based on a rhythmic phosphorylation of KaiC, and histidine protein kinases relay temporal information from the KaiABC complex to regulate gene expression. The kinases SasA and CikA engage directly with the oscillator and are responsible for modulating the phosphorylation and dephosphorylation throughout the circadian day of the response-regulator transcription factor RpaA; the phosphorylation state of RpaA in turn determines circadian gene expression. We recently showed that either CikA or SasA can drive rhythmic phosphorylation and DNA binding of RpaA in an in vitro system. However, when SasA is absent in vivo, a bioluminescence reporter gene shows a very low expression and amplitude rhythm, indicating CikA kinase activity is not sufficient to activate gene expression. We questioned why CikA cannot serve as a robust kinase for RpaA in the absence of SasA in the cell. Here, we investigated post-translational modifications of CikA and found KaiC-dependent phosphorylation sites of CikA that dramatically affect its activity. Phosphomimetic mutants of these sites showed that the phosphorylated version of CikA is not functional. Our data show that inverse correlation of KaiC levels and these inhibitory phosphorylation sites can explain the lower CikA activity in a SasA knockout background. We conclude that these phosphorylation sites act as a rheostat for CikA activity and are regulated by KaiC levels.
Keywords
Circadian clocks are self-sustained oscillators that provide an internal sense of time for individual cells and/or whole organisms. Post-translational modifications are known to regulate the activity of clock components. A striking example is the phosphorylation of the animal clock protein Period by casein kinase 1 epsilon, which affects clock function from Drosophila to humans. In addition, expressing different phosphomimetic mutant variants of the core mammalian circadian oscillator protein CRY1 produces changes in period length or arrhythmic gene expression (Kloss et al., 1998; Liu and Zhang, 2016; Maywood et al., 2014; Toh et al., 2001). In the Neurospora crassa fungal circadian clock model, single-site phosphomimetic substitutions in the White Collar-2 protein affect White Collar Complex-dependent transcription and circadian period length (Sancar et al., 2009). And in plants, phosphorylation by casein kinase 2 inhibits the interaction of the key circadian oscillator protein, CCA1, with its target gene promoters (Yan et al., 2021). Hence, regulation of clock components by post-translational modification is fundamental to biological timing.
Synechococcus elongatus PCC 7942 (hereafter S. elongatus) is a well-studied model organism for a cyanobacterial circadian clock and is known for maintaining circadian phosphorylation rhythms of its clock components in a test tube (Nakajima et al., 2005). The S. elongatus core oscillator consists of three proteins named KaiA, KaiB, and KaiC, and rhythmic phosphorylation/dephosphorylation of KaiC is the critical time keeper (Fang et al., 2024). Input and output pathway kinases SasA and CikA are responsible for phosphorylation (Takai et al., 2006) and dephosphorylation of the response-regulator RpaA (Gutu and O’Shea, 2013), which is the key transcription factor to drive S. elongatus rhythmic gene expression (Markson et al., 2013). The 24-h cycle of phosphorylation of KaiC, when combined with KaiA, KaiB, and ATP in a test tube, has provided a rich system to study the dynamics and structure of the time-keeping mechanisms. A classic model of circadian gene expression, based on genetic data, holds that KaiA stimulates the auto-kinase activity of KaiC that induces its phosphorylation (Iwasaki et al., 2002; Williams et al., 2002). In turn, phosphorylated KaiC stimulates SasA kinase activity, and SasA relays the time information by phosphorylating the response-regulator RpaA. Once phosphorylated, RpaA activates the expression of many downstream genes. However, when KaiC is fully phosphorylated, it provides a binding site for active KaiB to interact with the Kai complex; KaiB stimulates the auto-phosphatase activity of KaiC and recruits CikA, an interaction that activates CikA phosphatase activity on RpaA (Tseng et al., 2017). Implicit in this model is the necessity of SasA to phosphorylate, and CikA to dephosphorylate, RpaA, creating the rhythmic wave of active RpaA.
Missing from this model is the evidence that CikA, when it is not associated with the Kai complex, is a better kinase/phosphatase for RpaA than is SasA (Chavan et al., 2021). Moreover, either CikA or SasA can alone provide rhythmic output from the Kai complex in vitro, with CikA catalyzing RpaA phosphorylation in the day portion of the cycle and actively dephosphorylating RpaA in the night, when CikA is associated with KaiBC. SasA appears to provide only daytime kinase activity, with a slower RpaA autodephosphorylation. These data support the role of CikA as a kinase for the circadian clock in vivo without the requirement of SasA.
The current study aimed to reconcile in vitro and in vivo data. Consistent with the classic model, deletion of CikA results in elevated pkaiBC::luc reporter activity with small nighttime troughs (Mutsuda et al., 2003); this result is expected from continued daytime SasA kinase activity on RpaA and the absence of CikA phosphatase activity in the night. However, in the absence of SasA, the pkaiBC::luc reporter shows low levels of low-amplitude activity (Iwasaki et al., 2000). Until now, it has not been clear why CikA cannot serve as the sole kinase/phosphatase to generate high-amplitude rhythms of RpaA phosphorylation in the absence of SasA in vivo as it does in vitro. In this study, we found that CikA activity is inhibited in vivo by phosphorylation when KaiC levels are low or absent, as is the case in SasA or KaiC knockout backgrounds.
Materials and Methods
Immunoprecipitation and Mass Spectrometry of FLAG-CikA
Cultures used for IP were grown under a 2-day 12:12 light/dark cycle to entrain and then released to constant light. In initial experiments, cells were collected at CT12 (12 h after lights on) for immunoprecipitation (IP) and tandem mass spectrometry (LC-MS/MS) as described below and reported in Figure 1a and Table S1. Based on these results, for further experiments, cells were collected at CT4 (4 h after lights on) and CT22 (22 h after lights on) from the strains indicated in the figure legends and frozen in liquid nitrogen (reported in Figure 4). Cells were lysed by vortexing in TAP buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 0.1% Triton-X, 0.5 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail tablet Roche, REF11836170001) and glass beads (Sigma, G9143). The clearing centrifugation step was done for 10 min at 15,000 rpm at 4 °C. Supernatants were collected. Meanwhile, FLAG-M2 magnetic beads (30 µl per sample) were washed with 5 column volumes (CV) of Tris-buffered saline (TBS, 50 mM Tris pH 8.0, 100 mM NaCl) 3 times and then equilibrated with 5 CV of TAP buffer 3 times. The cleared supernatant was incubated with FLAG-M2 magnetic beads at 4 °C on a rotating wheel for 4 h. The FLAG-M2 magnetic beads were then washed with 5 CV of TAP buffer 3 times and 5 CV of TBS until the TBS absorbance at 280 nm was less than 0.05. The FLAG-M2 magnetic beads were stored at −80 °C. Afterwards, protein samples were diluted in TNE (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid [EDTA]) buffer. RapiGest SF reagent (Waters Corp.) was added to the mix to a final concentration of 0.1%, and samples were boiled for 5 min. Tris(2-carboxyethyl)phosphine (TCEP) was added to 1 mM (final concentration), and the samples were incubated at 37 °C for 30 min. Subsequently, the samples were carboxymethylated with 0.5 mg/ml of iodoacetamide for 30 min at 37 °C, followed by neutralization with 2 mM TCEP (final concentration). Protein samples prepared as above were digested with trypsin (trypsin:protein ratio = 1:50) overnight at 37 °C. RapiGest was degraded and removed by treating the samples with 250 mM HCl at 37 °C for 1 h, followed by centrifugation at 14,000 rpm for 30 min at 4 °C. The soluble fraction was then transferred to a new tube, and the peptides were extracted and desalted using C18 desalting columns (Thermo Scientific, PI-87782). Peptides were quantified using a Bicinchoninic Acid protein assay, and a total of 1 µg of peptides were injected for LC-MS analysis.
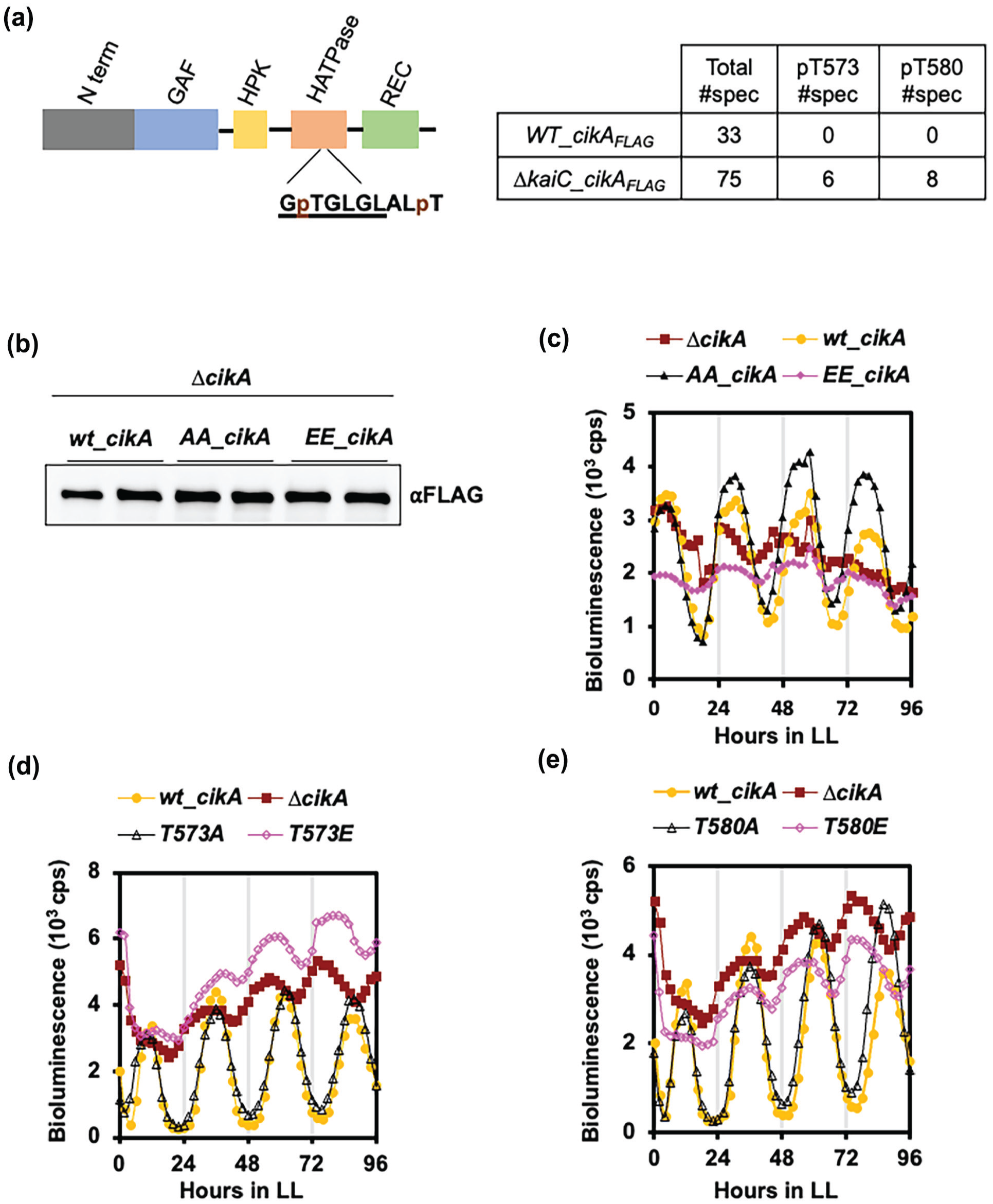
A phosphomimetic mutant of CikA cannot substitute for wt_CikA. (a) Scheme showing the conserved domains of CikA (left panel). Amino acids in the conserved G-2 box are underlined. Phosphorylated threonines (T573 and T580), identified by LC-MS/MS from samples collected at CT12, are labeled with “p.” Peptides that include phosphorylated threonines are shown in the table for the WT and ∆kaiC backgrounds (right panel). #Spec indicates the number of peaks that represent the unique ionized fragments in the spectrum of the peptide samples that include T573 and/or T580. (b) Western blot showing the expression levels of FLAG-tagged wt_CikA, AA_CikA, and EE_CikA for two independent clones in the ∆cikA background expressed by the native cikA promoter from the NS1 ectopic cloning site. (c) Bioluminescence reporter assay showing the expression of PkaiBC::luc under constant light for ∆cikA, wt_cikA, AA_cikA, and EE_cikA strains. Each trace represents the average of two different clones (n = 12). (d) Bioluminescence reporter assay showing the expression of PkaiBC::luc under constant light for ∆cikA, wt_cikA, T573A, and T573E strains. Each trace represents the average of two different clones (n = 12). (e) Bioluminescence reporter assay showing the expression of PkaiBC::luc under constant light for ∆cikA, wt_cikA, T580A, and T580E strains. Each trace represents the averages from two independent clones (n = 12).
Tandem Mass Spectrometry
Trypsin-digested peptides were analyzed by ultra-high-pressure liquid chromatography (UPLC) coupled with LC-MS/MS using nanospray ionization. The nanospray ionization experiments were performed using an Orbitrap Fusion Lumos hybrid mass spectrometer (Thermo) interfaced with a nano-scale reversed-phase UPLC (Thermo Dionex UltiMate 3000 RSLC nano System) using a 25-cm, 75-micron ID glass capillary packed with 1.7-µm C18 (130) BEH beads (Waters Corporation). Peptides were eluted from the C18 column into the mass spectrometer using a linear gradient (5%-80%) of acetonitrile (ACN) at a flow rate of 375 µl/min for 1.5 h. The buffers used to create the ACN gradient were Buffer A (98% H2O, 2% ACN, 0.1% formic acid) and Buffer B (100% ACN, 0.1% formic acid). Mass spectrometer parameters were as follows: an MS1 survey scan using the Orbitrap detector (mass range [m/z]: 400-1500 [using quadrupole isolation], 120,000 resolution setting, spray voltage of 2200 V, ion transfer tube temperature of 275 °C, Automatic Gain Control target of 400,000, and maximum injection time of 50 ms) was followed by data-dependent scans (top speed for most intense ions, with charge state set to only include +2-5 ions, and 5-second exclusion time, while selecting ions with minimal intensities of 50,000, during which the collision event was carried out in the high-energy collision cell (HCD Collision Energy of 30%), and the fragment masses were analyzed in the ion trap mass analyzer (with an ion trap scan rate of turbo, first mass m/z was 100, AGC target 5000, and maximum injection time of 35 ms). Protein identification was carried out using Peaks Studio 8.5 (Bioinformatics Solutions Inc.). MS analysis was performed at the “Molecular Mass Spectrometry Facility, UC San Diego.”
Protein Extraction
S. elongatus PCC7942 cell pellets were collected into 2-ml tubes and immediately frozen at −80 °C. For the total protein extraction, cell pellets were taken on ice and immediately mixed with TBS (50 mM Tris pH 8.0, 100 mM NaCl) containing 1 mM PMSF and a protease inhibitor cocktail (Roche, REF11836170001). Then, an appropriate amount of glass beads (Sigma, G9143) was added to the tube, and the mixture was vortexed for 10 min at 4 °C, followed by centrifugation at 15,000 rpm for 10 min at 4 °C. The supernatant was collected as the total protein extract, and protein concentrations were determined by Bradford colorimetric assay. For SDS-PAGE, the total protein extract was boiled at 95 °C for 5 min after adding 2X Laemmli sample buffer (BioRad catalog number 161-0737). For the Phos-tag sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), the total protein extract was mixed with 2X Laemmli sample buffer and either kept on ice for immediate loading or stored at −80 °C until loaded on the Phos-tag gel.
Western Blot Analysis
Total protein extract from S. elongatus PCC7942 cells was separated by SDS-PAGE and transferred to a nitrocellulose membrane (BioRad Cat #16220112) using the semi-dry transfer method with the BioRad Trans-Blot Turbo Transfer System according to the manufacturer’s instruction manual (Cat #1704150). The membrane was blocked with 5% non-fat milk in TBS (20 mM Tris-HCl pH 7.5) for 1 h at room temperature. FLAG antibody-decorated blots were incubated with Sigma-Aldrich monoclonal anti-FLAG M2-peroxidase (HRP) antibody (Cat #A8592) for 2 h at room temperature, followed by three 10-min washes with TBS-T (20 mM Tris-HCl pH 7.5, 0.1% Tween 20). The signal was developed using ThermoFisher SuperSignal West Femto Maximum Sensitivity Substrate (Cat #34095) and detected using a BioRad ChemiDoc Touch System.
Purification of GST-Tagged CikA and Mutants of CikA
The gene encoding CikA from S. elongatus was amplified using PCR and cloned in-frame with the PreScission Protease (GE Healthcare) cleavage site into the pET41a(+) vector (Novagen) between the NcoI and HindIII sites. The resulting plasmid was used to transform Escherichia coli BL21(DE3). Transformed E. coli cultures in log phase in LB medium at 37 °C were cooled to room temperature for 1 h and induced to overexpress recombinant CikA with 0.1 mM isopropyl β-D-thiogalactopyranoside (IPTG). Cells were harvested after 16 h, and pellets were resuspended in lysis buffer (50 mM Tris-HCl pH 7.5 with 150 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT) and then lysed using an Avestin Emulsiflex C3 homogenizer (Avestin Inc). The cell lysate was centrifuged at 15,000 rpm for 1 h at 4 °C. The GST-fusion CikA-containing supernatant was loaded onto GSTrap FF 5-ml columns pre-equilibrated with 5 CV of lysis buffer. Following GST-fusion CikA binding to the column, the bound material was washed with 5 CV of lysis buffer. The column was then equilibrated with 5 CV of PreScission Protease buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 8.0). Overnight PreScission protease digestion was performed at 4 °C, and the following day, untagged CikA was eluted from the column with elution buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 8.0). Eluted CikA proteins were separated with a size-exclusion column (HiLoad 16/600 Superdex 200 pg code 28-9893-35) equilibrated with elution buffer. The final protein concentration was determined by Bradford colorimetric assay.
In Vitro Phosphorylation Assay of RpaA
Purified 0.65-µM wt-CikA and 2.5-µM RpaA, 0.65-µM AA-CikA and 2.5-µM RpaA, 0.65-µM EE_CikA and 2.5-µM RpaA were incubated in in vitro reaction buffer (20-mM Tris-HCl pH 8.0, 150-mM NaCl, 5-mM MgCl2, 1-mM ATP, 0.5-mM EDTA) at 30 °C for 1 h. Only 2.5-µM RpaA in in vitro reaction buffer was used as control. After 1 h, 7 µL from each reaction was immediately mixed with 2x Laemmli buffer and loaded on a pre-prepared Phos-tag gel for further western blot analysis.
Partial Trypsin Digestion
Recombinant WT_CikA and double-mutant variants (AA_CikA, EE_CikA) were purified from E. coli cells. The purified wt-CikA and double-mutant variant proteins (20 µL of ~0.2 mg/mL of each) were incubated with 5 µL of 1 mg/mL, 0.33 mg/mL, 0.11 mg/mL, 0.04 mg/mL, 0.012 mg/mL, and 0.004 mg/mL trypsin in PBS separately at 22 °C for 30 min. Protease activity was stopped by adding 6.25 µL of 5x SDS stop solution sample buffer containing 5.2 mM PMSF and 5.2 mM EDTA. Afterwards, samples were boiled at 95 °C for 5 min and loaded on a precast gel (BioRad Cat #4569036) immediately. After SDS-PAGE, gels were stained with InstantBlue Coomassie Protein Stain (ab119211) for 2 h and washed with water 3 times for 10 min before imaging. The image was taken with the BioRad ChemiDoc Touch System.
Microscopy
Indicated S. elongatus cells were grown at constant light with an intensity of ~30 µE m−² s−¹ for 3 days in liquid BG-11 medium without added antibiotics. Samples of cultures were spotted on 1% (w/v) agarose in BG-11 medium covered with a coverslip. Cells were imaged using a Delta Vision inverted epifluorescence microscope (Applied Precision). Images were captured using a CoolSnap HD charge-coupled device (CCD) camera (Photometrics). Cell length measurements were obtained from TIFF images using ImageJ software (National Institutes of Health) (Schneider et al., 2012). Statistical analysis was carried out using Microsoft Excel.
Bioluminescence Reporter Assay
Flask-grown cultures were diluted to a density of OD750 = 0.05 and grown under constant light until reaching a density of OD750 = 0.2. Then, 20 µL of each culture was mixed with 10 µL of 100-mM firefly luciferin and placed on a Grenier Bio-One 96-well microplate (Cat #655073) filled with BG-11 agar medium. Plates were covered with clear tape to prevent drying, and holes were poked using a sterile needle to allow air transfer. Cultures were synchronized by incubating the plate under 12 h:12 h light (~30 µE m−² s−¹ light)/dark conditions for 2 days and then returned to constant light conditions for bioluminescence sampling. Bioluminescence of PkaiBC::luc firefly luciferase fusion reporter was monitored at 30 °C under constant light using an Infinite Pro M200 (TECAN). Data were analyzed using Microsoft Excel. Period analysis was performed on the BioDare2 website (biodare2.ed.ac.uk) (Zielinski et al., 2014).
Results
CikA Is Phosphorylated at Thr573 and Thr580 When KaiC Is Absent
While deletion of CikA regulates circadian phase resetting, period length, and cell size in S. elongatus (Miyagishima et al., 2005; Schmitz et al., 2000), not much is known about how CikA activity is regulated. We hypothesized that post-translational modifications of CikA could be important for its activity. To discover potential clock-dependent post-translational modifications of CikA, we fused an N-terminal FLAG epitope to CikA, producing a FLAG-tagged variant that complemented a cikA deletion mutant (Supplemental Figure 1A). FLAG-tagged CikA was immunoprecipitated by FLAG-M2 magnetic beads in reporter strain AMC541 (hereafter WT) and its ∆kaiC deletion derivative and then analyzed by mass spectrometry. We found robust phosphorylation at Thr573 and Thr580 sites specifically in the absence of KaiC (Figure 1a, Supplemental Table 1). Thr573 lies in an HATPase domain within the conserved G2-box motif of CikA, whereas Thr580 resides just after the G2-box, suggesting potential regulatory roles for these phosphorylation sites. To test the effect of lower KaiC levels on the phosphorylation of these sites, we repeated the experiment using a sasA deletion strain in which KaiC levels are low compared to WT (Takai et al., 2006). As with the kaiC deletion mutant, the sites were phosphorylated in the sasA deletion but not in the WT background (Supplemental Figure 1C right panel), consistent with KaiC dependence of phosphorylation. CikA interaction with core components is low in the daytime, and when CikA is mutated on C644 to arginine (C644R), its ability to interact with KaiBC is decreased (Tseng et al., 2017). Hence, in an independent experiment, we used a WT background carrying FLAG-tagged WT CikA or its C644R variant at CT4 (early day) when KaiABC-CikA interaction is expected to be low, and at CT22 (late night) when KaiABC-CikA interaction is expected to be high. T580 phosphorylation was detected with WT CikA at CT4 when CikA-KaiABC interaction is low (Supplemental Figure 1D, left panel). In addition, we detected more phosphorylation of the C644R mutant on both the T573 and T580 sites at CT4, supporting the notion that higher phosphorylation at these sites occurs when CikA interaction with core clock components is diminished (Supplemental Figure 1D, right panel). Together, these data suggest that phosphorylation of CikA Thr573 and Thr580 sites is induced when CikA is not interacting with core clock components, either due to affinity for or levels of core clock components.
Phosphomimetic Variants of CikA Cannot Substitute for WT CikA
To investigate the importance of CikA Thr573 and Thr580 phosphorylation sites on CikA function, we exchanged both sites to alanine (AA_CikA) to prevent phosphorylation of CikA, and to glutamate (EE_CikA) to mimic phosphorylated CikA. Alleles that expressed FLAG-tagged WT_CikA, AA_CikA, or EE_CikA from the native cikA promoter were expressed in a ∆cikA strain to characterize their resulting phenotypes. AA_CikA and EE_CikA were both present at levels similar to WT_CikA as analyzed by western blot using the anti-FLAG antibody (Figure 1b). Moreover, expressing AA_CikA or EE_CikA did not affect the expression levels of KaiC compared to WT_CikA (Supplemental Figure 2C). However, the allele expressing the phosphomimetic variant EE_CikA did not complement the cikA deletion phenotype in a circadian bioluminescence reporter assay (Figure 1c). The EE_cikA mutant also produced elongated cells under low light growth, similar to the ∆cikA strain (Supplemental Figure 2A and 2B). These data suggest that phosphorylation of CikA at T573 and T580 hinders its activity, and hence, the phosphorylated mimic EE_CikA cannot fulfill CikA’s role in circadian clock gene regulation or in controlling cell size. Mutation of either the T573 or T580 site to glutamate resulted in same cikA null phenotype for the bioluminescent reporter (Figure 1d and 1e), indicating that single phosphorylation at this region is enough to inhibit CikA activity. Similarly, mutation of either T573 or T580 to alanine was sufficient to produce CikA’s robust gene expression, as observed in the double mutant (Figure 1c-1e).
Kinase Activity of EE_CikA on RpaA Is Diminished Due to a Possible Conformation Change
CikA is both a kinase and phosphatase for RpaA (Gutu and O’Shea, 2013). Our in vivo data suggest that the EE_CikA variant produces a cikA-deletion phenotype because it cannot relay the circadian signal to RpaA. To investigate the kinase activity of CikA phospho-variants, we purified WT_CikA, AA_CikA, EE_CikA, and RpaA from E. coli and performed in vitro phosphorylation assays. When purified RpaA was incubated with WT_CikA or AA_CikA, both CikA variants induced RpaA phosphorylation as analyzed by Phos-tag gel (Figure 2a). However, EE_CikA was not able to phosphorylate RpaA (Figure 2a). These data indicate that EE_CikA does not have kinase activity for RpaA.
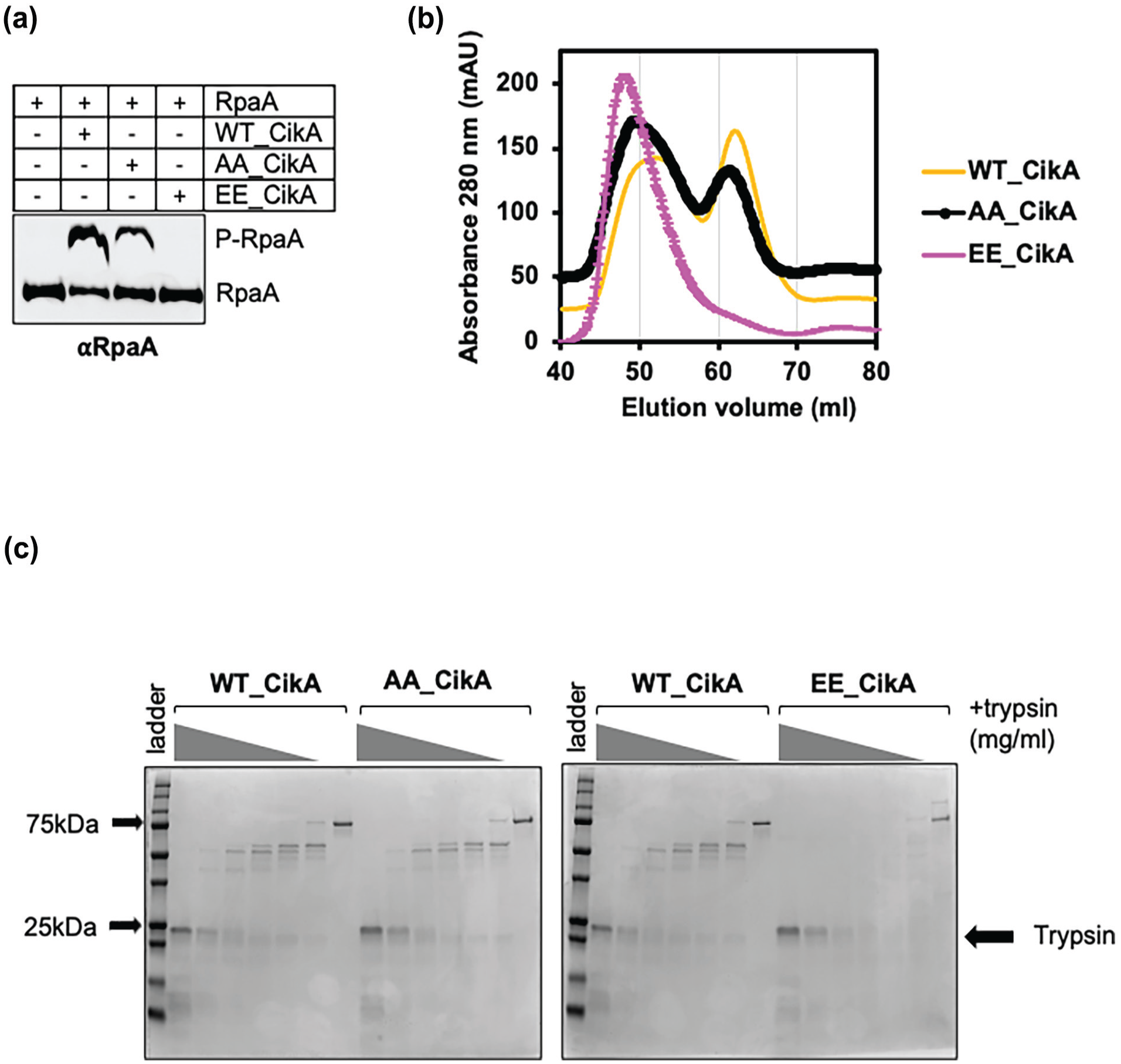
Kinase activity of EE-CikA on RpaA is diminished due to a possible conformation change. (a) Western blot showing the phosphorylated and unphosphorylated form of purified RpaA from Phos-tag SDS-PAGE after incubation with WT_CikA, AA_CikA, or EE_CikA, respectively. (b) Combined size-exclusion column elution graphs showing the WT_CikA, AA_CikA, and EE_CikA elutions. (c) Coomassie-stained SDS-gels showing partial trypsin digestion of CikA variants. Purified CikA variants incubated with trypsin at 1 mg/mL, 0.33 mg/mL, 0.11 mg/mL, 0.04 mg/mL, 0.012 mg/mL, 0.004 mg/mL, and 0 mg/mL.
These phosphorylation sites are around the G2-box, which forms a flexible loop in histidine kinases (Bilwes et al., 1999). To test whether phosphorylation at this region induces a conformational change, we performed size-exclusion chromatography of purified CikA variants. WT_CikA and AA_CikA eluted as two peaks, whereas EE_CikA eluted as a single high-molecular-weight peak, which suggests that EE_CikA has a different conformation compared to WT_or AA_CikA (Figure 2b). Due to elution in the high-molecular-weight range, we hypothesized that EE_CikA has a more open structure. To test this hypothesis, we utilized partial trypsin digestion to compare sensitivity of WT_CikA, AA_CikA, and EE_CikA to trypsin. EE_CikA was more sensitive to trypsin, consistent with a more open conformation than WT_CikA and AA_CikA (Figure 2c). In addition, purified EE_CikA ran slightly slower than WT_CikA or AA_CikA when analyzed by Phos-tag SDS-PAGE (Supplemental Figure 2D). Together, the data suggest that phosphorylation at Thr573 and Thr580 induces a conformational change on CikA that inhibits its kinase activity against RpaA.
AA_CikA Compensates for the Low-Amplitude Rhythm Observed in ∆sasA
A recent study showed that CikA can be a more robust kinase as well as phosphatase for RpaA than SasA in the presence of KaiABC in vitro (Chavan et al., 2021). However, a ∆sasA strain shows low-magnitude and low-amplitude (Supplemental Figure 3A) and short-period (Supplemental Figure 3B) bioluminescence rhythms compared to WT and has low KaiC levels (Taniguchi et al., 2010). We hypothesized that CikA is less active in ∆sasA due to low KaiC levels, which enables phosphorylation at Thr573 and/or Thr580. As a support for this hypothesis, we recently showed that the low-amplitude phenotype of a sasA-deletion strain can be rescued by elevating KaiC levels via expressing an additional copy of kaiBC (∆sasA_Ptrc::kaiBC) (Chavan et al., 2021). Moreover, the bioluminescence rhythm for pkaiBC::luc in a ∆sasA_Ptrc::kaiBC strain is dependent on CikA activity (Figure 3a). Conversely, increasing KaiB and KaiC in the absence of CikA cannot induce a robust bioluminescence rhythm in a sasA-deletion strain. Together, these data suggest that increased KaiC levels can rescue CikA from being phosphorylated and inactivated in the absence of SasA, thereby increasing the CikA activity on RpaA.
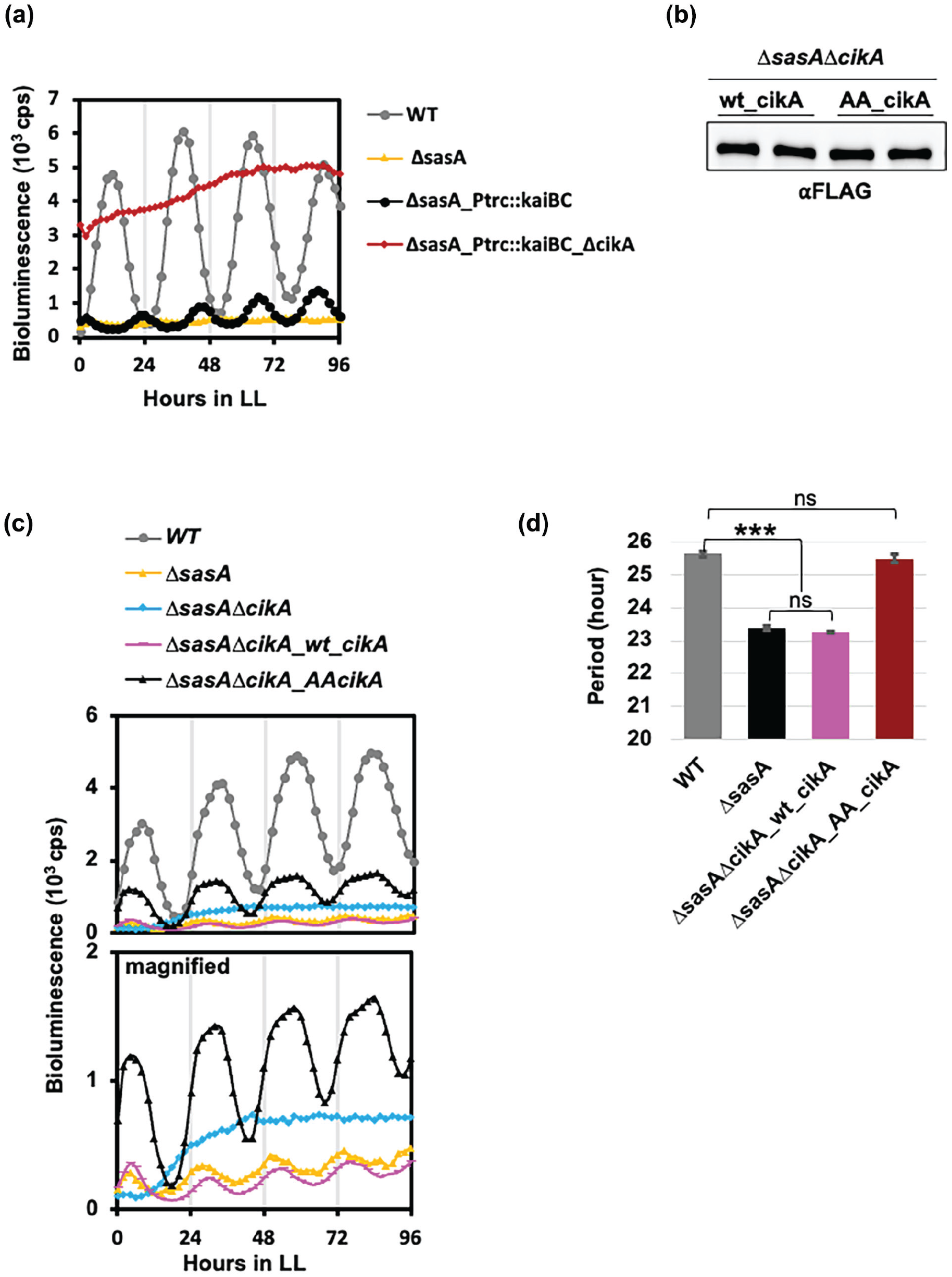
AA_CikA compensates for low-amplitude rhythm and short period observed in ∆sasA. (a) Bioluminescence reporter assay showing the expression of PkaiBC::luc under constant light for ∆sasA, ∆sasA_Ptrc::kaiBC, and ∆sasA_Ptrc::kaiBC∆cikA strains. Each trace represents the averages from two independent clones (n = 12). (b) Western blot showing the expression levels of FLAG-tagged wt_CikA and AA_CikA in ∆sasA∆cikA double-deletion background from two independent clones. (c) Bioluminescence reporter assay showing the expression levels of PkaiBC::luc under constant light for WT, ∆sasA, ∆sasA∆cikA, ∆sasA∆cikA_wt_cikA, and ∆sasA∆cikA_AA_cikA. Each trace represents the averages from two different clones (n = 12). (d) Bar graph showing the period lengths of WT, ∆sasA, ∆sasA∆cikA_wt_cikA, and ∆sasA∆cikA_AA_cikA. Data are represented as mean ± SEM (n = 6).
To test whether the low-amplitude gene expression rhythm in the ∆sasA mutant is due to phosphorylated and inactivated CikA, we expressed WT_CikA and AA_CikA in a ∆sasA∆cikA double-deletion background (Figure 3b). We predicted that the AA_CikA mutant should produce better rhythms because it cannot be phosphorylated at the inhibitory phosphorylation sites. Indeed, we observed higher-amplitude bioluminescence rhythms from the AA_CikA expresser compared to the WT_CikA expresser in the ∆sasA∆cikA double-deletion background (Figure 3c lower panel). Moreover, expressing AA_CikA in the ∆sasA∆cikA double-deletion background corrected the ~2-h short period observed in ∆sasA (Figure 3d). These data independently support our hypothesis that the unphosphorylated AA_CikA mutant is a more robust kinase and phosphatase for RpaA and show that this non-phosphorylatable CikA variant can overcome the defects of sasA deletion and serve as the sole output kinase for the circadian clock in vivo, as has been shown in vitro.
Time-Dependent Interaction Partners of CikA Require KaiABC Interaction
Our data suggest that CikA could form at least two different complexes; one on the KaiABC complex (hereafter, Kai complex) and the other not interacting with the Kai complex. In order to identify potential time-dependent CikA interaction partners, we analyzed the CikA complexes at CT4 (off the Kai complex) and CT22 (on the Kai complex). We identified 125 proteins that were retrieved with WT_CikA compared to a mock control (Figure 4a, upper panel) (Supplemental Table 2). Among these proteins, 43 interacted regardless of the time of the day, whereas 25 were enriched at CT4, and 57 were enriched at CT22 (Supplemental Table 2). We also used the C644R mutant, which binds poorly to the Kai complex, as a control to find out the contribution of CikA-KaiABC interaction on the CikA interactome. The total number of interaction partners was decreased for C644R, and especially for CT22-specific ones (Figure 4a, lower panel, Supplemental Figure 4B). CikA peptides detected by MS in different samples were comparable, minimizing the contribution of IP efficiencies on the detection of interaction partners (Supplemental Table 2). We analyzed the fold enrichment of 125 WT_CikA-interacting candidates in CT4 vs CT22 (-10lgP fold > 1.5) in WT_CikA and C644R experiments to see whether the C644R substitution changes time-specific interaction. Among the CT4-enriched group, the C644R substitution resulted in decreased interaction specifically at CT4 (Figure 4b, left panel, Supplemental Table 3). Interestingly, half of the CT22-enriched interaction partners still associated with C644R, albeit with loss of the time specificity (Figure 4b, right panel, Group 1, Supplemental Table 3). Nonetheless, half of CT22-specific interaction partners were lost with C644R (Figure 4b, right panel, Group 2). Only a few interaction partners of WT_CikA behaved similarly when C644R was used (Figure 4b, right panel, Group 3). Together, the data suggest that CikA-KaiABC interaction determines both the time and intensity of the majority of CikA interactions.
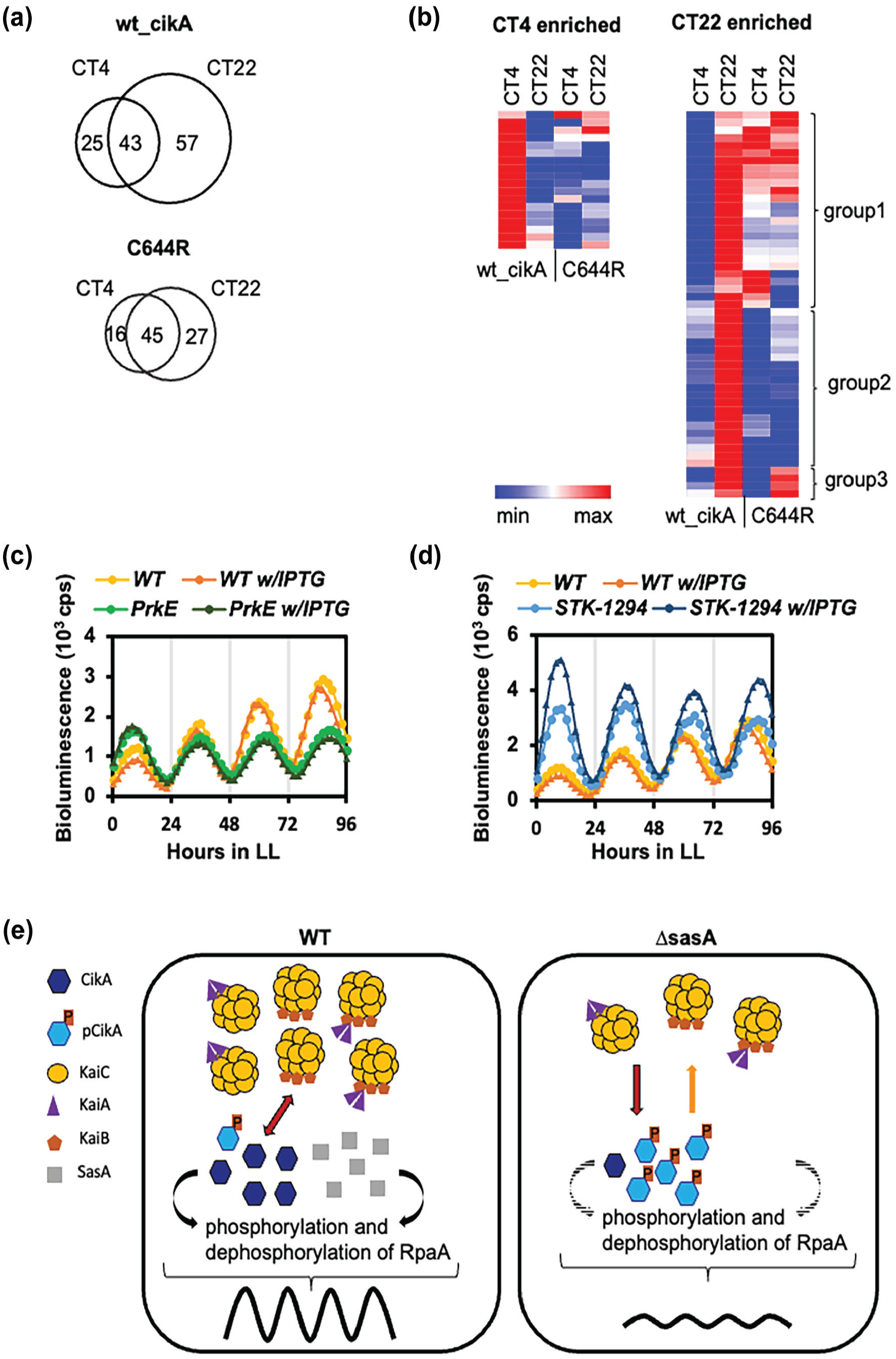
Time-dependent interaction partners of CikA are also dependent on clock interaction. (a) Venn diagrams showing the number of putative interaction partners of CikA with wt_CikA (upper panel) and the C644R mutant (lower panel) at CT4 and CT22. For stringent analysis, the potential interaction partners with the 50 > -10lgP value were excluded. (b) Heat map showing the significance (-10lgP) of the time-specific interaction partners of CikA. Interaction partners enriched in CT4 (>1.5 fold) and CT22 (>1.5 fold) are included in WT and C644R backgrounds. The color scale ranges from red to blue, where red indicates higher significance, and blue indicates lower significance of interaction. (c) Bioluminescence reporter assay showing the expression of PkaiBC::luc under constant light for WT and PrkE (WT_Ptrc::prkE) with and without IPTG. Each trace represents the average of two different clones (n = 12). (d) Bioluminescence reporter assay showing the expression of PkaiBC::luc under constant light for WT and STK-1294 (WT_Ptrc::STK-1294) with and without IPTG. Each trace represents the average of two different clones (n = 12). (e) Scheme showing the difference in rhythmic regulation of gene expression between WT and ∆sasA strains. Active SasA and CikA associate with the Kai complex at different times of day, stimulating them to phosphorylate/dephosphorylate the transcription factor RpaA, and generating rhythmic gene expression. In WT cells with sufficient KaiC and KaiB levels, CikA associates with the Kai complex when the KaiB ring has formed and is dephosphorylated and remains active, allowing for rhythmic gene expression (bidirectional arrow); the association of SasA with the Kai complex, maximal when the KaiB ring has not formed, is not depicted (left panel). In ∆sasA mutant cells with reduced KaiC and KaiB levels compared to WT (right panel), there is less association of CikA with the Kai complex (solid and hatched arrows); a significant portion of CikA is phosphorylated and inactive, resulting in reduced amplitude of rhythmic gene expression. Even though rhythmic gene expression is maintained, the magnitude of gene expression is much lower (reduced peaks, reduced amplitude) due to inactive CikA kinase and the absence of SasA-mediated phosphorylation of RpaA.
Among the putative interaction partners of CikA and C644R, we found two serine/threonine kinases (STK) as potential interactors of CikA: Synpcc7942_0600 (previously named PrkE; Mackey et al., 2008) and Synpcc7942_1294 (hereafter STK-1294). To test whether either PrkE or STK-1294 is responsible for CikA T573 and/or T580 inhibitory phosphorylation, we expressed them ectopically in S. elongatus under the control of an IPTG-inducible Ptrc promoter and with a FLAG tag. We hypothesized that if either (or both) of these kinases is responsible for these inhibitory phosphorylations, over-expressing them could decrease CikA activity. However, overexpression of neither PrkE nor STK-1294 (Supplemental Figure 4C) produced cikA- deletion phenotypes in bioluminescence rhythms (Figure 4c and 4d). The data suggest that either there are other kinases responsible for phosphorylation of these sites, or other proteins are required to generate a permissive state for CikA phosphorylation. Because non-phosphorylated CikA does not cause a mutant phenotype under normal conditions, a screen to identify an unknown kinase or other factors was not practical.
Discussion
Although CikA was shown to have a significant role in the cyanobacterial circadian clock almost 25 years ago, the biochemical regulation of its kinase and phosphatase activities is not well understood. Recent work has shown that the in vitro clock can guide in vivo experiments to yield deeper insights from mutant phenotypes than were gleaned during initial characterization. For example, a combination of in vitro and in vivo experiments recently explained why the circadian clock in a cikA mutant strain does not change its phase of gene expression in response to environmental resetting cues as a wild type does, even though the in vitro KaiA, KaiB, KaiC oscillator, lacking either kinase, resets the phase of KaiC phosphorylation, as does the intact WT cell (Fang et al., 2023). The surprising insight was that an in vitro clock built with SasA as the only output kinase present, similar to the cikA mutant, resists phase resetting. Conversely, a CikA-only in vitro clock is a “super-resetter.” Follow-up experiments to test the phase-response properties of a sasA mutant in vivo confirmed that, when CikA is present in the absence of SasA, the clock resets to an exaggerated degree. The very low residual bioluminescence signals from a sasA-deletion strain had deterred further experimentation until the in vitro experiments provided compelling hypotheses worth exploring.
Similarly, in vitro clock experiments challenged the interpretation of CikA’s role in the clock as a phosphatase, with SasA dominating the role as kinase. CikA in vitro is a potent kinase of RpaA, and it supports very robust rhythmic phosphorylation of RpaA in a clock reaction. We sought to answer why RpaA phosphorylation levels are so low, and rhythms are of such low amplitude, in the absence of SasA in vivo. One possible answer to this question was that CikA activity is reduced by cellular factors in a way that does not occur during the in vitro clock cycle. Here, we showed that CikA is inhibited by phosphorylation when KaiC levels are low. This mechanism unravels, at least partially, why CikA does not support strong rhythms of gene expression in vivo when SasA is absent. Low levels of KaiC in a sasA-deletion strain potentiate an unknown kinase that phosphorylates and inactivates CikA. Expressing the variant AA_CikA (T573A and T580A double mutants) that cannot be phosphorylated enhanced the circadian oscillation in a sasA-deletion background. In addition, expression of AA_CikA corrected the period-length phenotype, although amplitude did not reach WT levels.
Exchange of these phosphorylation sites to glutamate, either individually or in combination, rendered CikA inactive. Thr573 site resides inside, and Thr580 just after, the G2-box that acts as a nucleotide-binding motif in histidine kinases. Previous studies showed by mutation of the glycine residues that the G2-box plays an important role in modulating phosphatase and/or kinase activities (Tanaka et al., 1998; Zhu and Inouye, 2002). Our data indicate that a phosphomimetic (EE) version of Thr573 and Thr580 does not have any kinase activity against RpaA. Because the G2-box is where ATP binds, we hypothesize that phosphorylation at this site could interfere with ATP binding due to the ionic interactions with phosphorylated threonine and ATP. Another possible regulation could be a conformation change of CikA upon phosphorylation at these sites that affects partner interaction. This idea is supported by the higher sensitivity of EE_CikA to limited proteolysis. Previous studies focusing on the role of G2-box in histidine kinases focused on exchange of conserved glycine residues. S/T residues are found in the second position of many G2-boxes of histidine kinases in bacteria (Kim and Forst, 2001). Hence, it would be of interest whether histidine kinases in other organisms are regulated by inhibitory phosphorylation of G2-box sites.
CikA is proposed to interact with the Kai clock complex primarily during the night, and this interaction is decreased when CikA is mutated on the C644 site to Arg (C644R) (Tseng et al., 2017). When we compared the putative interaction partners of CikA during day (CT4) and night (CT22), we observed more interaction partners at night when CikA is on the Kai complex. The C644R mutant lost most of this discrimination between day and night putative interaction partners. In addition, when CikA affinity to the oscillator complex is decreased as in C644R, both the time and intensity of the interactions are affected, suggesting that the clock orchestrates the CikA interactome. Moreover, phosphorylation of T573 and/or T580 in the C644R_CikA mutant was greater in the day than in night, when CikA interaction with the core clock is more limited due to both circadian time and the C644R mutation. Together, these data support our hypothesis that CikA is selectively inactivated and, when phosphorylated, has a potential conformation change that prevents its interaction with partners, including RpaA. While the KaiABC/CikA/RpaA clock can function in vitro without the requirement of other feedback loops, this inhibitory phosphorylation of CikA could enhance the robustness of the clock by limiting CikA activity to the subpopulation that is interacting with the clock. In future, it would be of interest to use strains that carry the unphosphoryatable AA_CikA variant to determine whether selective CikA inactivation contributes to the robustness of the circadian clock.
Supplemental Material
sj-docx-1-jbr-10.1177_07487304251338156 – Supplemental material for Clock-Dependent Phosphorylation of CikA Regulates Its Activity
Supplemental material, sj-docx-1-jbr-10.1177_07487304251338156 for Clock-Dependent Phosphorylation of CikA Regulates Its Activity by Cigdem Sancar and Susan S. Golden in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-2-jbr-10.1177_07487304251338156 – Supplemental material for Clock-Dependent Phosphorylation of CikA Regulates Its Activity
Supplemental material, sj-xlsx-2-jbr-10.1177_07487304251338156 for Clock-Dependent Phosphorylation of CikA Regulates Its Activity by Cigdem Sancar and Susan S. Golden in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-3-jbr-10.1177_07487304251338156 – Supplemental material for Clock-Dependent Phosphorylation of CikA Regulates Its Activity
Supplemental material, sj-xlsx-3-jbr-10.1177_07487304251338156 for Clock-Dependent Phosphorylation of CikA Regulates Its Activity by Cigdem Sancar and Susan S. Golden in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-4-jbr-10.1177_07487304251338156 – Supplemental material for Clock-Dependent Phosphorylation of CikA Regulates Its Activity
Supplemental material, sj-xlsx-4-jbr-10.1177_07487304251338156 for Clock-Dependent Phosphorylation of CikA Regulates Its Activity by Cigdem Sancar and Susan S. Golden in Journal of Biological Rhythms
Footnotes
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM118290. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
The authors have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Considerations
This article does not contain any studies with human or animal participants.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.