
Abstract
Hedgehog (Hh) signaling plays an important role in embryonic patterning and adult stem cell renewal but has recently been found also to be involved in certain stem cell cancers. One of the first steps in Hh signaling is the autoprocessing of Hh protein, in which the C-terminal domain (Hh-C) catalyzes a cholesterol-dependent autocleavage reaction that leads to the production of the cholesterol ester of the N-terminal Hh domain (Hh-N), thereby yielding a signaling molecule that activates the Hh pathway by binding to the Patched receptor. This article describes an in vitro, homogeneous assay system that measures changes in fluorescence polarization that accompany the cholesterol-dependent autocleavage of Hh protein. The assay system makes use of a modified Hh protein in which Hh-N, which is not essential for autocleavage, is replaced by a 25-residue peptide containing a tetracysteine motif, complexed with a bisarsenical fluorophore. The assay is quite robust and easily adapted to high-throughput screening in 384-well plates with Z′ factors above 0.8. It has been used to screen the National Institutes of Health Clinical Collection, which has led to the identification of 2 compounds that inhibit the cholesterol-dependent autocleavage of Hh protein at micromolar concentrations.
Keywords
Introduction
H
Most research on the role of Hh signaling is focused on the series of events that lie between the activation of Smo and the activation of Gli, with the aim of understanding this complex signaling pathway and in the hope of developing potential cancer therapeutics. In contrast, very little attention has been given to the role of Hh autoprocessing and modification, despite the fact that it constitutes the initial step in the Hh signaling pathway. Owing to the absence of pharmacologic tools for its study, the role of Hh autocleavage and esterification with cholesterol in Hh signaling, both in the developing embryo and in the growth of Hh ligand-dependent cancers, is still not entirely clear.
All earlier studies on the autoprocessing of Hh protein used denaturing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) to follow the autocleavage reaction, 2,4-6 which does not lend itself easily to a rapid, quantitative assay. This article describes a high-throughput screen (HTS) for identifying compounds that attenuate Hh autoprocessing and can be used as research tools for dissecting the role of autoprocessing and the attendant cholesterol modification in Hh function, both in the many different contexts of embryonic development and in Hh ligand-dependent stem cell cancers. The HTS makes use of an in vitro, homogeneous assay system that measures changes in fluorescence polarization that accompany the cholesterol-dependent autocleavage of Hh protein. It involves replacing the signaling domain of Hh protein, which is not essential for autocleavage, with a 25-residue peptide containing a FlAsH-tag, which is complexed with a bisarsenical fluorophore based either on fluorescein (FlAsH) or resorufin (ReAsH). 7
The data presented in this article show that this assay is quite robust and easily adapted to HTS in 384-well plates with Z′ factors 8 above 0.8. The assay has been used to screen a small compound library and has led to the identification of 2 compounds that inhibit the cholesterol-dependent autocleavage of Hh protein at micromolar concentrations.
Materials and Methods
Chemicals and reagents
4′,5′-bis(1,3,2-dithioarsolan-2-yl)-fluorescein (FlAsH) and 4′,5′-bis(1,3,2-dithioarsolan-2-yl)-resorufin (ReAsH) as the 1,2-ethanedithiol (EDT) complexes were synthesized as described 7,9 or purchased from Invitrogen (Carlsbad, CA). The National Institutes of Health (NIH) Clinical Collection was obtained from BioFocus (South San Francisco, CA)
Plasmid construction
The tetracysteine motif, CCPGCC or FlAsH tag, was inserted into the inducible coding region of plasmid pET45b (Novagen, Madison, WI) adjacent to the His-tag, using the Kpn I and NotI restriction sites and appropriate synthetic oligonucleotides (5′-GTGTTGCCCGGGTTGCTGTGC-3′ and 5′-GGCCGCACAGCAACCCGGGCAACACGTAC-3′), custom synthesized by Invitrogen, to yield plasmid pET45H. A plasmid encoding the hedgehog protein of Drosophila melanogaster was generously provided by Dr. Kent Nybakken of this institute. The entire hedgehog coding sequence was amplified by the polymerase chain reaction using Failsafe polymerase (Epicentre, Madison, WI) and the primers 5′-GGCGGCCGCTTCCCACGTGCACGGCTG-3′ and 5′-GGCGGCCGCTCGTTGGGATCAGGATCGCTC-3′ (Invitrogen). The amplification product was cloned into PCR-XL-TOPO (Invitrogen), the recombinant plasmid was digested with NotI restriction endonuclease, and the 0.71-kb fragment containing the hedgehog coding sequence was inserted into the NotI site of plasmid pET45H to yield plasmid pET45Dmel. The predicted amino acid sequence of the 26.3-kDa hedgehog protein with its N-terminal His-tags and FlAsH-tags is shown in

Deduced amino acid sequence of the recombinant Hh protein expressed by plasmid pET45Dmel. The His-tag and FlAsH-tag sequences are underlined, the site of autocleavage is indicated by the vertical arrow, and start of the autoprocessing domain, with the N-terminal sequence, CFT, is indicated.
Bacterial growth and protein expression
Escherichia coli Rosetta 2 (DE3) (Novagen), transformed with plasmid pET45Dmel, was cultured in Luria-Bertani (LB) medium supplemented with chloramphenicol (34 µg/mL) at 37 °C. In mid-exponential phase (A600 = 0.5), isopropyl β-D-thiogalactopyranoside was added to a final concentration of 0.4 mM, and the cells were harvested by centrifugation 3 h later, washed once with Buffer A (20 mM Na phosphate [pH 7.5], 0.5 M NaCl, 1 mM p-toluenesulfonyl fluoride), and stored at −70 °C.
The bacterial cells were thawed, suspended in Buffer A (3 mL per 50 mL of culture), and disrupted in a French pressure cell at 4 °C. The extract was centrifuged at 20,000 g for 30 min, and the pellet was washed twice with Buffer A and extracted twice with 3 mL of Buffer B (20 mM Na phosphate [pH 7.5], 0.5 M NaCl, 8 M urea, and 1 mM p-toluenesulfonyl fluoride) to solubilize inclusion bodies. SDS-PAGE analysis of the solubilized inclusion bodies showed the major protein component to be the 26-kDa hedgehog protein. The inclusion bodies were stored at 4 °C but could also be frozen at −80 °C without significant loss of autoprocessing activity.
Fluorescence polarization assay for Hh autoprocessing
The assay was typically carried out in black 384-well plates (Nunc 264558) with 70 µL per well and was initiated by mixing equal volumes of a solution containing 1.4 µM (40 µg/mL) of solubilized inclusion bodies diluted in 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 0.4 mM 2-(dimethylamino)ethanethiol, and 0.7 µM FlAsH-EDT2, in Buffer D (20 mM Na phosphate [pH 6.9], 0.5 M NaCl, and 0.5 M L-arginine hydrochloride) with a solution of 68 µM cholesterol in 0.04% Triton X-100. In larger assays, liquid dispensing was done with a Thermo Fisher Multidrop 384 dispenser (Thermo Fisher, Waltham, MA). The mixtures were incubated at 25 °C, and fluorescence polarization was measured at 10-min intervals in a Tecan Safire 2 microplate reader (Tecan, Männedorf, Switzerland) with excitation at 390 nm and emission at 550 nm. A time delay of 300 ms was imposed between plate movement and flash to eliminate a fluorescence polarization (FP) artifact caused by vibration of the meniscus, which can give rise to systematic errors in FP values. To measure only the first step in the Hh autoprocessing reaction involving formation of the thioester intermediate (
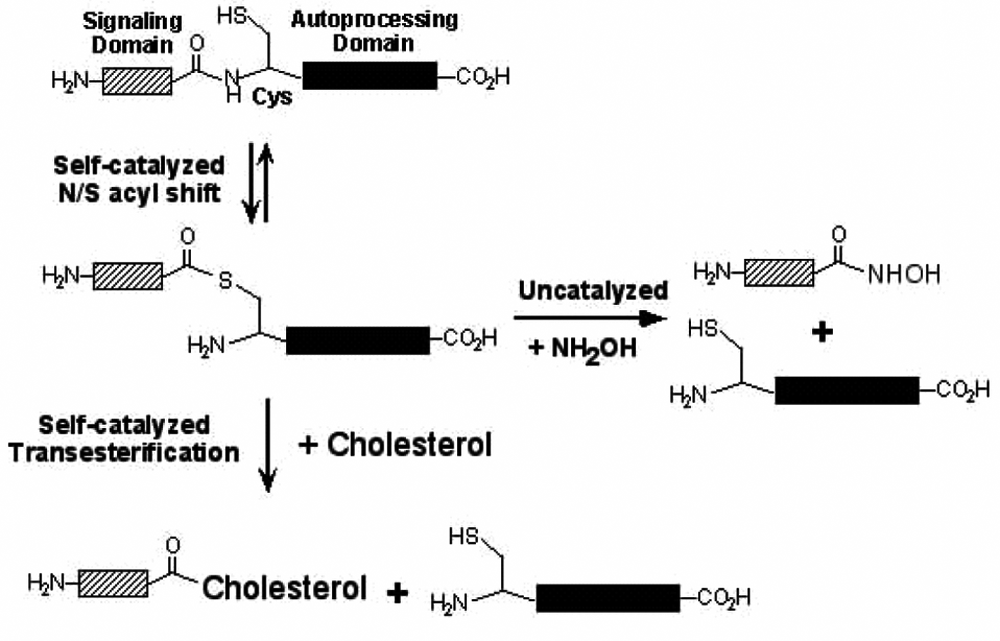
Mechanism of hedgehog (Hh) autoprocessing. The first step in autoprocessing is a self-catalyzed rearrangement of the peptide bond at the N-terminus of the autoprocessing domain to a thioester involving the side chain of its conserved N-terminal Cys residue. This is followed by transesterification to cholesterol, bound to the C-terminal autoprocessing domain, to yield the cholesterol ester of the signaling domain, which functions in intracellular signaling. Alternatively, the thioester intermediate can be cleaved by a strong nucleophile such as hydroxylamine.
Results
Assay development
Hh autoprocessing is a 2-step reaction, the first step being the N/S acyl rearrangement of the scissile bond between the signaling and the autoprocessing domains and the second the transesterification of the resulting ester intermediate with cholesterol (
Our assay takes advantage of the fact that Hh autoprocessing requires only the C-terminal autoprocessing domain and that the N-terminal signaling domain can be replaced with any peptide,
2
for example, a 25-residue peptide that contains a tetracysteine motif (FlAsH-tag), as shown in
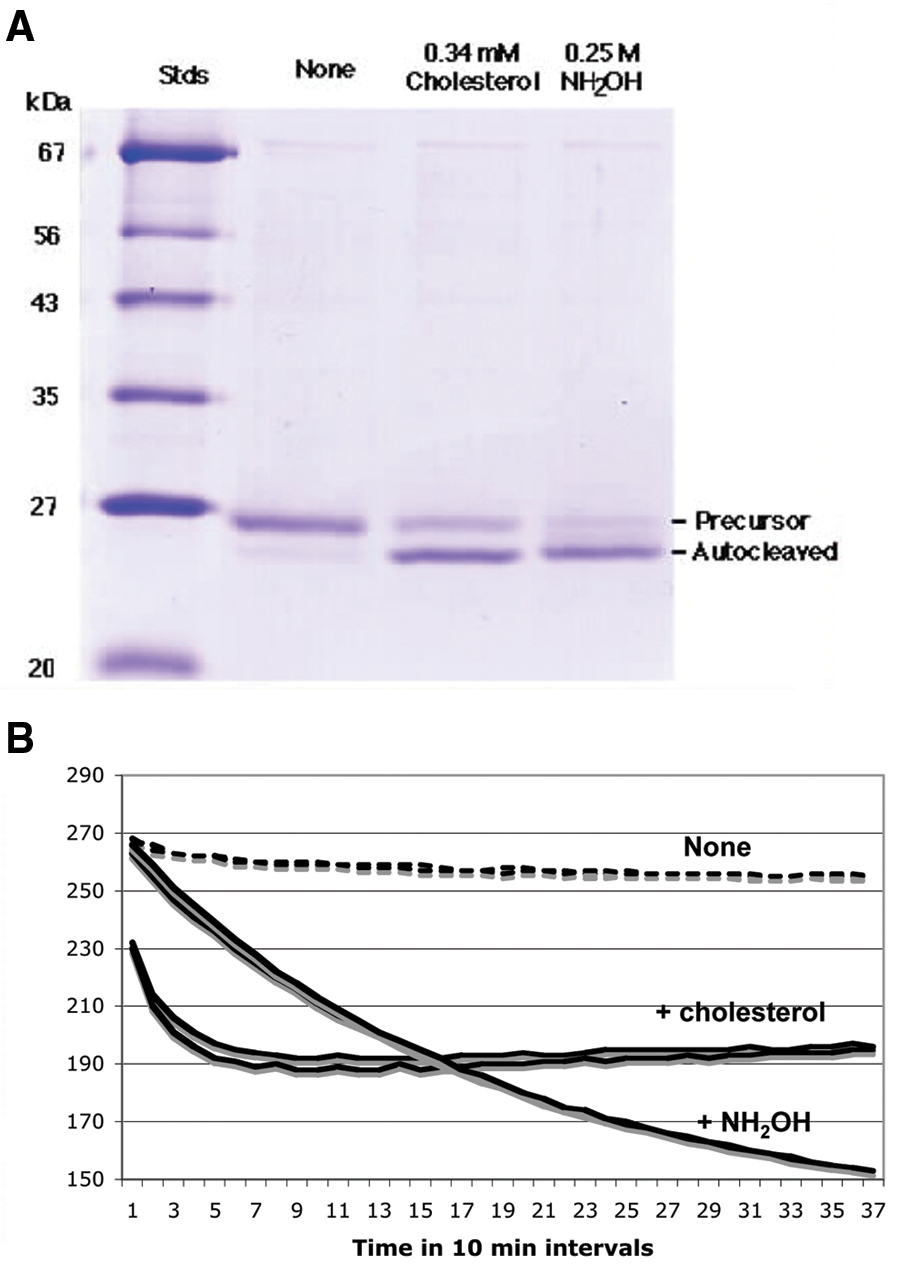
Autoprocessing of the HhC fusion protein with an N-terminal FlAsH-tag. (
Monitoring of the autoprocessing reaction by fluorescence polarization (FP) using ReAsH. FP was measured in triplicate wells of a 384-well plate upon excitation at 590 nm and emission at 632 nm, without nucleophile or with either 34 µM cholesterol or 2.2 M hydroxylamine, using 1 µM Hh protein and 4.4 µM ReAsH-EDT2, which had been dialyzed after complex formation to remove unbound bisarsenical fluorophore.
Critical parameters and further assay optimization
The rate and extent of reaction of FlAsH-tagged peptides with FlAsH-EDT2 depend on reagent concentration as well as on the presence of low concentrations of monothiols, 9 which makes the relative concentrations of these components critical factors in the FP polarization assay. TCEP (1 mM) was added to ensure full reduction of the Cys residues of the FlAsH-tag, and 2-(dimethylamino)ethanethiol (0.4 mM) was used to promote complex formation between the FlAsH-tag and the bisarsenical fluorophore. Under these conditions, FlAsH-bisarsenical complex formation went to completion in about an hour at 25 °C, requiring a preincubation period prior to running the FP assay. This made for a relatively reproducible procedure and was used in our pilot screen of the NIH Clinical Collection (see below) but may lead to some plate-to-plate variation in the context of a large HTS. Moreover, the presence of fluorescent impurities in some batches of FlAsH-EDT2 or ReAsH-EDT2 required careful optimization of the relative concentrations of the FlAsH-tagged protein and each lot of bisarsenical fluorophore.
We subsequently developed a more reproducible procedure for fluorescent labeling of the FlAsH-tagged Hh-C protein and the bisarsenical fluorophore, which used a molar excess of the fluorophore to induce complex formation, followed by dialysis against buffer containing 1 mM TCEP but no free thiol to remove unbound fluorophore. This dialyzed mixture gave reproducible assays over a period of 4 days and is therefore well suited for large-scale HTS efforts. The data shown in
Another important assay parameter is the manner of dispersion of the water-insoluble cholesterol substrate. We examined a number of detergents, of which octyl-β-D-glucopyranoside and Triton X-100 gave the most robust cholesterol-dependent activities. Triton X-100 was the preferred reagent because it gave stable cholesterol dispersions slightly above the critical micelle concentration (CMC) but could be diluted in the assay to 0.2%, which was just below the CMC and had no effect on autoprocessing activity but was still high enough to minimize “promiscuous” inhibition.
11
Under these conditions, the apparent Km for cholesterol was about 14 µM, compared to a Km of 29 µM for the cholesterol analog, ergosterol (
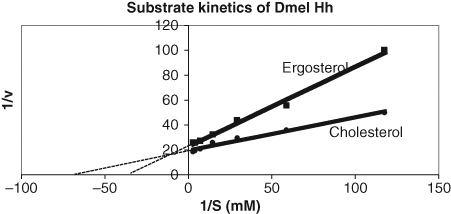
Substrate kinetics of the autoprocessing of the HhC fusion protein with an N-terminal FlAsH-tag. The assay was conducted as described in the legend to
Pilot screen of the NIH Clinical Collection
To evaluate the suitability of our FP assay for HTS, we conducted a pilot screen of the NIH Clinical Collection. The 446 compounds of this library were assayed in duplicate at 25 µM, using the FlAsH-based assay in 384-well plates. Every assay plate contained at least 2 rows of negative controls without inhibitor and 2 rows of positive controls in which 100% inhibition was simulated by omitting the substrate cholesterol. The assay was conducted as described in
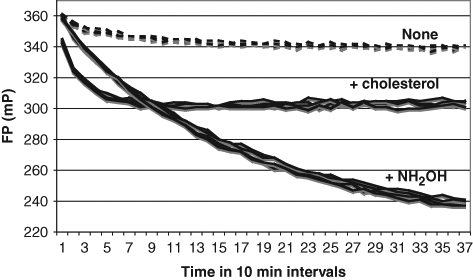
The 2 hits with >50% inhibition in the pilot screen were subjected to secondary screening using the resorufin-based bisarsenical reagent, ReAsH, with excitation at 590 nm and emission at 632 nm, to rule out any interference of the compounds with the fluorescence measurements at 550 nm used in the primary screen. The secondary screen measured concentration dependence and also compared cholesterol- and hydroxylamine-dependent autocleavage. As shown in
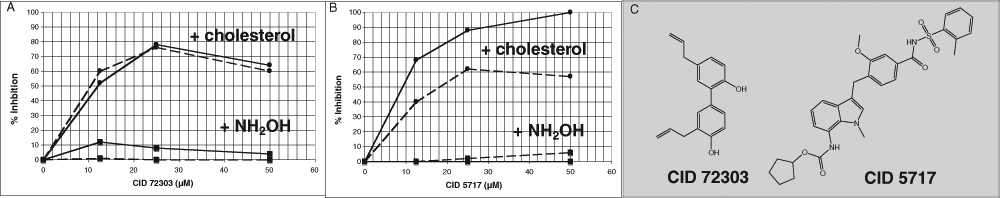
Secondary screening of inhibitors identified in the pilot screen. The 2 compounds from the National Institutes of Health Clinical Collection that gave more than 50% inhibition at 25 µM were assayed at the concentrations indicated either in the presence of 34 µM cholesterol in Triton X-100 dispersion or 2.2 M hydroxylamine as an artificial nucleophile, as indicated on the chart. Fluorescence polarization (FP) was measured in a 384-well plate upon excitation at 590 nm and emission at 632 nm, using 2 µM Hh protein and 0.4 µM ReAsH-EDT2, either after 1 h (solid lines) or 4 h (dotted lines). (
Discussion
The Hh signaling pathway is the subject of intense investigation, owing to its important role in embryonic development and certain types of cancer. 1,3 However, most current research focuses on the steps that occur after the Hh ligand binds to the patched receptor (Ptch1). In contrast, the steps by which the Hh ligand is generated from its precursor form, which include 3 different processing events 10 and interaction with at least 2 additional proteins prior to binding to Ptch1, 12,13 have not been studied in detail, not least owing to the lack of chemical probes for their dissection. Until the recent identification of a small molecule, robotnikinin, which binds to the sonic Hh signaling domain, 14 the only probes available for studying the early steps in the Hh pathway had been antibodies that bind to Hh-N and block its activity. 15 The assay described in this article represents the first attempt to identify inhibitors of the self-catalyzed autocleavage reaction, which is coupled to the conjugation of the Hh signaling domain with cholesterol. Such inhibitors would be valuable tools for investigating the role of Hh autocleavage and esterification with cholesterol in Hh signaling, about which there is still some uncertainty because catalytically impaired Hh mutants have significant residual function, 4 and artificial Hh-N variants without lipid modification can function in some signaling contexts (e.g., Fujita et al. 16 ).
Our FP assay relies on the reduction of anisotropy as the fluorophore-labeled N-terminal protein segment is reduced in size from 27 kDa to 3 kDa by hedgehog protein autoprocessing. When autoprocessing is induced by hydroxylamine as an artificial nucleophile, the change in FP is nearly 50% (
When applied to the 446-compound NIH Clinical Collection using the FlAsH-based assay, our HTS behaved in a robust manner with only 2 positives, defined by more than 50% inhibition at 25 µM, which were confirmed by dose-dependent secondary assays, using the resorufin-based fluorophore, ReAsH, as an indicator to eliminate potential fluorescence artifacts in the primary screen (
Footnotes
Acknowledgements
The authors thank Drs. Charles P. Emerson, Kent Nybakken, and Xing-bin Ai for helpful discussions; Dr. Anton Simeonov (NIH/NHGRI) for many valuable suggestions; and Dr. Kent Nybakken for a gift of the plasmid encoding the hedgehog protein of Drosophila melanogaster.
This work was supported by a grant from the Concern Foundation (Xing-bin Ai) and by grant R21 NS064844 from the National Institutes of Health (Henry Paulus and Charles P. Emerson).