
Abstract
Nek2 is a serine/threonine protein kinase that localizes to the centrosome and is implicated in mitotic regulation. Overexpression of Nek2 induces premature centrosome separation and nuclear defects indicative of mitotic errors, whereas depletion of Nek2 interferes with cell growth. As Nek2 expression is upregulated in a range of cancer cell lines and primary human tumors, inhibitors of Nek2 may have therapeutic value in cancer treatment. The authors used a radiometric proximity assay in a high-throughput screen to identify small-molecule inhibitors of Nek2 kinase activity. The assay was based on the measurement of the radiolabeled phosphorylated product of the kinase reaction brought into contact with the surface of wells of solid scintillant-coated microplates. Seventy nonaggregating hits were identified from approximately 73,000 compounds screened and included a number of toxoflavins and a series of viridin/wortmannin-like compounds. The viridin-like compounds were >70-fold selective for Nek2 over Nek6 and Nek7 and inhibited the growth of human tumor cell lines at concentrations consistent with their biochemical potencies. An automated mechanism-based microscopy assay in which centrosomes were visualized using pericentrin antibodies confirmed that 2 of the viridin inhibitors reduced centrosome separation in a human tumor cell line. The data presented show that pharmacological inhibition of Nek2 kinase results in the expected phenotype of disruption to centrosome function associated with growth inhibition and further supports Nek2 as a target for cancer drug discovery.
Introduction
M
Nek2 is a serine/threonine protein kinase that is regulated in a cell cycle–dependent manner. 2 It is the closest relative in the human genome of the NIMA kinase of Aspergillus nidulans, which is an essential regulator of mitotic progression. Nek2 is activated by dimerization and autophosphorylation and inhibited through interaction with and dephosphorylation by protein phosphatase 1. 3 Nek2 is localized to the centrosome, where it regulates spindle pole separation at the onset of mitosis through phosphorylation and displacement of proteins, including C-Nap1 and rootletin. 3 There is also evidence that it contributes to chromatin condensation and spindle checkpoint function. 3
Nek2 is abnormally expressed in cancer cells. 4 Initially, microarray studies revealed increased expression of Nek2 mRNA in Ewings tumor cell lines and diffuse large B cell lymphomas. Subsequently, elevated levels of Nek2 protein have been identified in a wide variety of cancer cell lines as well as in a significant proportion of primary human cancers, including breast tumors, cholangiocarcinomas, and testicular seminomas. 5-8 The mechanism for upregulation of Nek2 expression remains to be determined. However, the locus that carries the Nek2 gene, 1q32, is amplified in both breast and gastric tumors. 9,10
Experimental studies suggest that abnormal Nek2 expression may contribute to the classic tumor hallmarks of aneuploidy and chromosome instability. Overexpression of active Nek2 leads to premature centrosome separation and the accumulation of cells with multiple nuclei and supernumerary centrosomes, whereas overexpression of kinase-inactive Nek2 or depletion by RNAi of the wild-type enzyme interferes with centrosome separation and bipolar spindle formation. 6,11,12 These data support the hypothesis that Nek2 activity is carefully regulated in normal cells to promote accurate cell division. Importantly, total Nek2 depletion in HeLa cells results in the arrest of cell proliferation, raising the possibility that Nek2 inhibitors might block cancer progression. 13 Also, RNAi-based depletion of Nek2 selectively interfered with the proliferation of cholangiocarcinoma cell lines but not normal fibroblast cell lines and led to a reduction in tumor size and peritoneal dissemination of cholangiocarcinoma tumor xenografts. 7 Meanwhile, RNAi knockdown of Nek2 in ER-positive and ER-negative human breast cancer cell lines reduced cell growth and migration and the size of human breast tumor xenografts. 8
Although an inhibitor of the interaction of the spindle checkpoint protein, Hec1, with Nek2 has been described, 14 no selective inhibitors of Nek2 kinase activity have been reported. The purpose of this work was to identify in a nonbiased high-throughput screen small-molecule inhibitors of Nek2 kinase activity. Here we describe the outcome of the screen and the chemical properties and cellular effects of some of the compounds identified.
Materials and Methods
Materials and Km determination are described in supplementary material.
FlashPlate® assay
384-Well FlashPlates were coated overnight at 4°C with myelin basic protein (MBP; 25 µg/mL in phosphate-buffered saline [PBS]), washed twice with PBS on a 16-pin plate washer (ELX50; Biotek Instruments Ltd., Northstar, UK), and compound (3 µL) in 2% DMSO added to each well (final nominal concentration 32 µM in 0.3% DMSO), followed by master mix (8 µL) consisting of buffer (50 mM HEPES pH 7.4 ± 0.2, 5 mM MnCl2, 5 mM β-glycerophosphate, 5 mM NaF, 10 mM MgCl2, 1 mM dithiothreitol [DTT]) with or without active Nek2 enzyme (20 pg/well). Finally, a mix of 10 µM adenosine triphosphate (ATP) and 0.2 µCi γ-33P-ATP (8 µL) was added (total volume 19 µL). These additions were made using a MiniTrak™ 5 (PerkinElmer Life Sciences, Waltham, MA). Plates were shaken (~1 min), incubated at room temperature (4 h), washed twice (10 mM sodium pyrophosphate), and then counted on a Topcount-NXT™ (PerkinElmer Life Sciences). Plates were formatted to contain total activity controls (n = 32), positive controls (staurosporine at 25 µM, n = 8), and no-enzyme blanks (n = 24) on the outside 2 columns either side of the plate. Signal-to-noise (S/N) and signal-to-background (S/B) ratios were calculated using the equations S/N = mean S – mean B/√(sdS)2 + (sdB)2 and S/B = mean S/mean B, respectively where sd = standard deviation. The Z′ factor was used to assess assay performance. 15 Screening data were analyzed using ActivityBase version 5.2 (IDBS, Guildford, UK). The activity of hits was confirmed in triplicate. Potency (IC50) was determined over a concentration range of 0.07 to 159 µM.
Filter assay
The kinase reaction buffer was as described above. Compounds (10 µM) were added to a 96-well polypropylene microplate, followed by substrate (10 µL of phospholemman [PLM] peptide at 20 µM) and ATP (10 µL of 20 µM) containing 0.25 µCi γ-33 P-ATP and the reaction initiated by adding Nek2 (20 pg/well; 50 µL volume). The plate was incubated at room temperature (90 min) and the reaction stopped by the addition of 2% orthophosphoric acid (30 µL). The solution was transferred to the wells of a prewetted phosphocellulose filter plate and the contents of each well filtered and washed twice with orthophosphoric acid (0.5%). Microscint 20 scintillant was added to each well (25 µL) and the plate read for 30 s per well using the Topcount-NXT™.
Kinase specificity assays
Specificity assays were performed using modification of an established method. 16 Recombinant kinases (1 µg/mL) were mixed on ice with compounds dissolved in DMSO or DMSO alone (2.5% DMSO final concentration) in a microplate. Kinase buffer (KB; 50 mM HEPES, KOH [pH 7.4], 5 mM MgCl2 [for Cdk1, Plk1, and Aurora A] or MnCl2 [for Nek2, Nek6, and Nek7], 5 mM β-glycerophosphate, 5 mM NaF, 4 µM ATP, 1 mM DTT, 20 nCi γ32P-ATP/mL) containing 0.5 mg/ml β-casein (for Nek2, Nek6, Nek7 and Plk1) or histone H1 (for Cdk1 and Aurora A) as substrate was added and the components mixed on ice by gentle pipetting. The plate was sealed and transferred to a water bath (30°C) for 30 min and the reactions stopped at 4°C by the addition of 0.5 M EDTA containing 0.2% bromophenol blue. Reaction products were slot-blotted as arrays onto nitrocellulose transfer membrane (Whatman, Maidstone, UK), washed 3 times in blotting buffer (25 mM Tris, 192 mM glycine, 10% MeOH (v/v)) and air-dried, exposed to a storage phosphoscreen (60 min), and analyzed using a Cyclone Phosphorimager and Optiquant analysis software (PerkinElmer Life Sciences). IC50 values calculated based on 2 independent experiments performed in triplicate using Prism 4.00 software with the bottom constraint set to zero (GraphPad Software Inc., San Diego, CA).
Cell culture and growth inhibition assays
The human cancer cell lines HeLa, U2OS, and tetracycline Nek2-inducible U2OS:GFP-Nek2A cells were cultured and induced as previously described. 11 For growth inhibition assays, cells were seeded (2.5-6 × 106 cells per well) into 96-well plates. Compounds (dissolved in DMSO) at a range of concentrations or DMSO (1%-2% final concentration) alone were added after 24 h and the cells incubated for 72 h. Cell numbers were measured using WST-1 reagents or sulphorhodamine B (SRB). The concentration of inhibitor that reduced cell growth by 50% (GI50) of DMSO control was calculated.
Immunofluorescence microscopy
Cells were fixed and processed for indirect immunofluorescence microscopy, 11 using rabbit antipericentrin (1:1000; Abcam, Cambridge, UK), mouse anti-γ-tubulin (1:500; Sigma, St. Louis, MO), and Alexa Fluor 594 goat antimouse or Alexa Fluor 488 goat antirabbit (1 µg/mL; Molecular Probes, Eugene, OR) antibodies. DNA was stained with Hoechst 33258 (0.2 µg/mL; Calbiochem, San Diego, CA) or DAPI (0.1 µg/mL; Invitrogen, Carlsbad, CA).
Automated cell-based assay for Nek2 activity
Tetracycline-inducible U2OS:GFP-Nek2A cells were seeded (4000 cells/well) in a black clear-bottomed plate and cultured overnight. Nek2A expression was induced with 1 µg/mL doxycycline (100 µL per well). PBS was added to the control (uninduced) wells. Simultaneously, test compound in 1% DMSO (v/v) final concentration or 1% DMSO alone was added and incubated overnight. Cells were washed once with PBS, fixed for 10 min on ice with cold 100% methanol (−20°C), and the methanol was removed by washing (3 times with PBS). An antibody to the centrosomal marker, pericentrin, was added (1.3 µg/mL in 5% bovine serum albumin [BSA]/PBS) and the plate incubated with gentle agitation for 1 h at room temperature and washed as before. Goat antirabbit Alexa-488 (2 µg/mL) antibody and the nuclear stain DAPI (0.1 µg/mL) were added in 5% BSA/PBS and the plate incubated for 1 h at room temperature, then washed (3 times PBS) and refrigerated until ready to image. Plates were read on an IN Cell Analyser 1000 (see supplemental material for details of image acquisition). An algorithm was designed to identify the DAPI-stained nuclei and to segment the cells setting a radius of 10 µm as the cell outline or “collar.” The pericentrin-identified centrosomes present only within the 10-µm cell radius were counted by adding another segmentation parameter (organelles). The size of the objects was optimized to be between 0.5 and 10 µm, with a pixel intensity (or sensitivity) set to 70. A threshold filter was added to the algorithm to classify the number/percentage of cells that had 1 or >1 centrosome.
Results
Determination of Km for ATP
The Km of ATP for Nek2A kinase was determined. Kinase reactions were performed using baculovirus-expressed Nek2A, β-casein as substrate, and buffer conditions as previously optimized.
17
By varying the ATP concentration and determining the initial rates of reaction, the Km for ATP was calculated to be 13.1 µM (
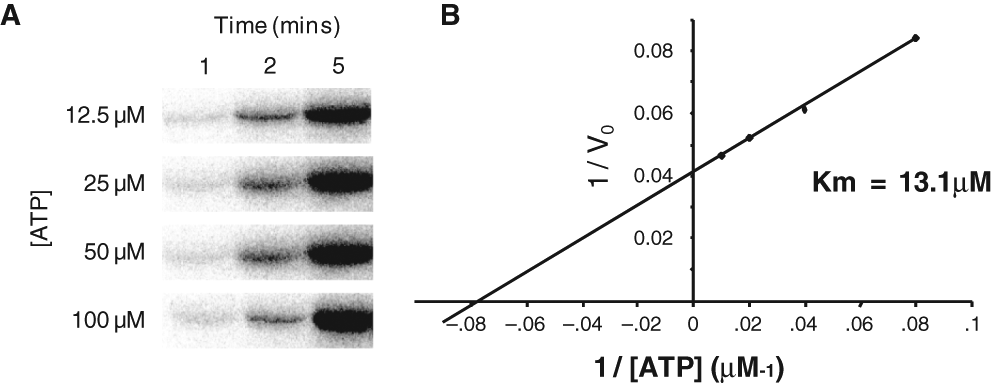
Determination of the Km for adenosine triphosphate (ATP) for the Nek2 kinase. (
Optimization of high-throughput screen for Nek2 inhibitors
A FlashPlate assay was designed as described in the Materials and Methods. MBP (25 µg/mL) provided a higher signal compared to either β-casein or phospholemman (PLM) peptide (data not shown). Also as expected, increasing the amount of unlabeled ATP (1-8 µM) competed with radioactively labeled ATP, thereby reducing the extent to which MBP was radioactively labeled and thus the signal obtained. A concentration of 2 µM ATP (plus 0.2 µCi γ-33P-ATP) was used in subsequent assays. The assay was DMSO tolerant up to 0.3% final concentration. Enzymatic linearity was observed for up to 4 h (S/B = 40) using 20 ng/well Nek2 (
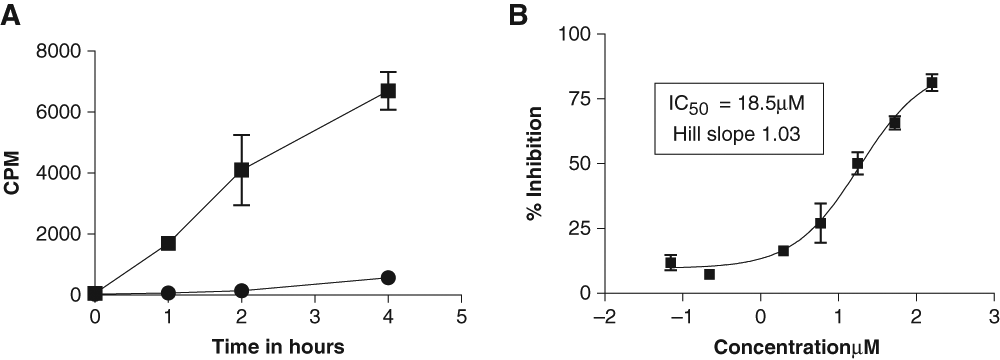
Optimization of conditions for Nek2 kinase biochemical assay. The conditions for the FlashPlate assay were established according to the Materials and Methods. (
High-throughput screen for inhibitors of Nek2 kinase activity
The overall S/B and S/N ratios for the library screen were 68 and 4.7, respectively. The Z′ factor was used to pass or fail each plate: plates were retested if the Z′ factor was <0.5. In a typical batch of 19 plates (
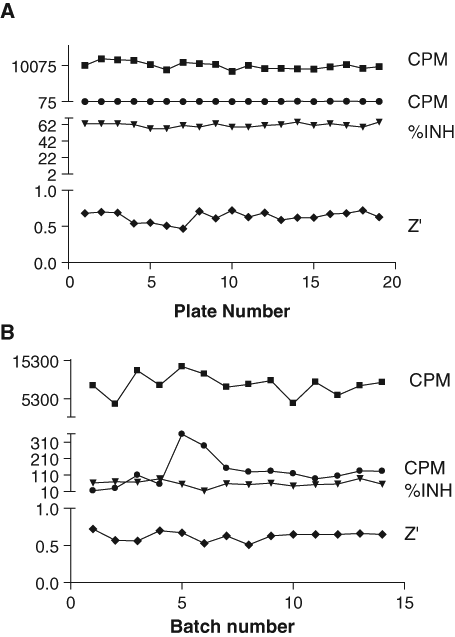
Assay performance achieved in the high-throughput screen. (
Compounds that inhibited enzymatic activity by >50% were selected as hits. The activity of 169 compounds was confirmed in triplicate (0.23% hit rate). Confirmed hits were screened to determine if the compounds were likely to be inhibiting Nek2 nonselectively via aggregation. 19 The assay used was identical to that described for the primary screen except that 0.01% Triton X-100 was added to the buffer. Seventy of the confirmed hits retained activity in this assay and were therefore likely to be nonaggregators. It should be noted, however, that the aggregating properties of early stage compounds may depend not only on the protein target but also on the type and concentration of the detergent used and the assay format, 20 and thus some compounds with aggregating properties may not have been identified. The potency (IC50) of these 70 compounds ranged from 0.65 to 91 µM.
Several series of compounds were identified as Nek2 inhibitors. These included a group of steroid-like viridins and another of toxoflavins. The chemical structures of the toxoflavins are shown in
Chemical Structures of 5 Toxoflavin Compounds Identified as Potent Inhibitors of Nek2 in High-Throughput Screening
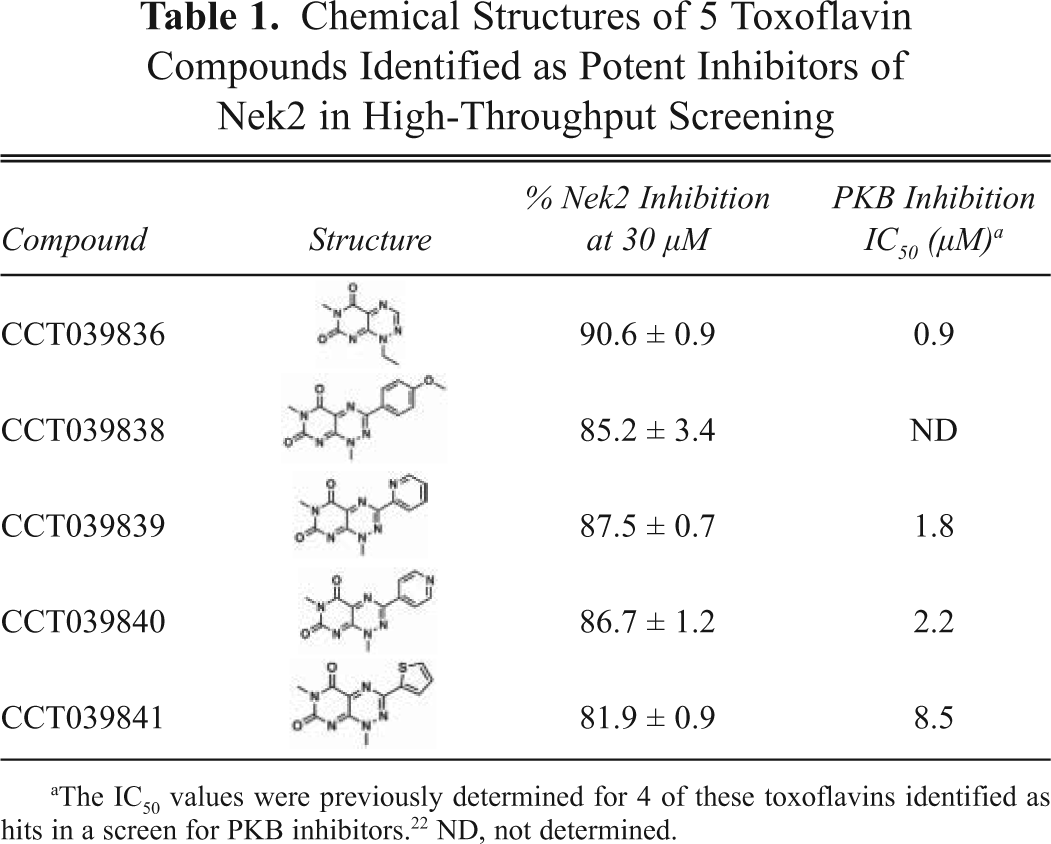
The IC50 values were previously determined for 4 of these toxoflavins identified as hits in a screen for PKB inhibitors. 22 ND, not determined.
The activity of a selection of the viridin-like compound series was confirmed in both the FlashPlate (IC50 = 0.5-2.9 µM) and the filter assay (IC50 = 1.4-11.9 µM;
Chemical Structures, Nek2 Kinase IC50 Values, and Antiproliferative Effects (GI50) of the Viridin-Like Compounds
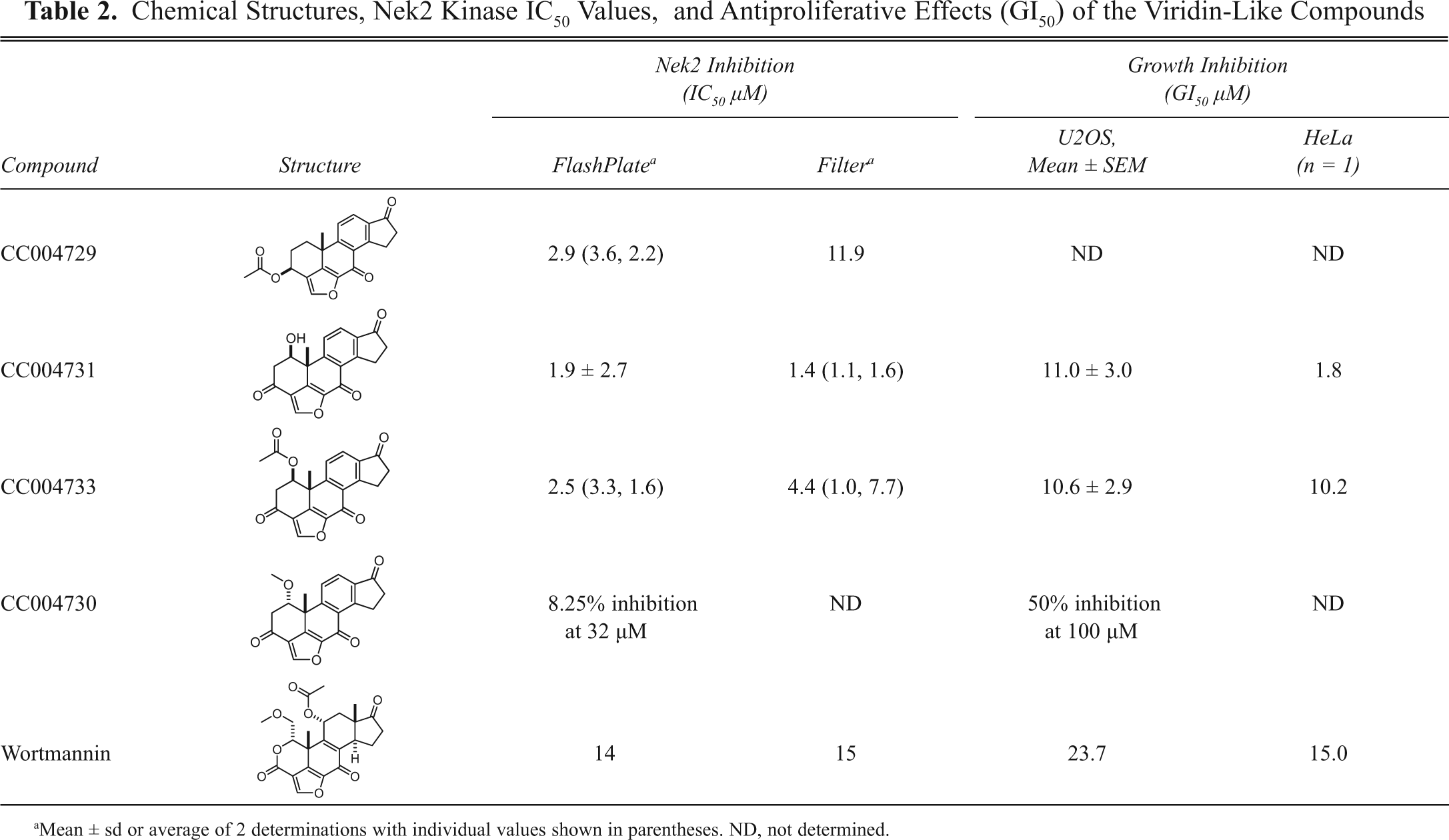
Mean ± sd or average of 2 determinations with individual values shown in parentheses. ND, not determined.
In addition, small molecules from a kinase-focused compound set were also identified as Nek2 inhibitors and their activities confirmed in the FlashPlate (IC50 = 11-47 µM) or the filter (IC50 = 7.3-39 µM) assays. Those hits (MW = 295-355, ClogP 1.1-4.1) did not contain potentially reactive chemical functionality and included a 2-amino pyridine, 4-(2-amino-5-(thiophen-3-yl)pyridin-3-yl)benzamide. This compound (
Selectivity of viridin compounds for mitotic kinases
Although the viridin-like compounds are known to inhibit members of different kinase families, we were specifically interested to compare their activity against Nek2 with their activity against other kinases that are implicated in mitosis.
3
Interestingly, the 2 viridin-like compounds, CC004731 and CC004733, showed a 70- to 1000-fold selectivity for Nek2 compared with Nek6 and Nek7 in this assay (
Selectivity of Viridin Compounds for Mitotic Kinases

IC50 values ± standard error (logEC50) were determined for staurosporine, wortmannin, CC004731, and CC004733 for the mitotic kinases Nek2, Nek6, Nek7, Aurora A, Plk1, and Cdk1/Cyclin B in a biochemical kinase assay. >400 µM denotes no inhibition at 400 µM inhibitor, the highest concentration used in the assay.
Antiproliferative effects of the viridin-like compounds
Nek2 depletion using RNAi approaches results in an antiproliferative phenotype.
7,8,13
Both CC004731 and CC04733 caused growth inhibition of the osteosarcoma cell line U2OS (GI50 = 11.0 and 10.6 µM, respectively;
Cell-based assay for Nek2 inhibition
The most well-characterized role of Nek2 is in promoting centrosome separation at the onset of mitosis. 2 Under normal cell cycle conditions, the activation of Nek2 at the G2/M transition triggers the loss of cohesion between the duplicated interphase centrosomes by phosphorylating and displacing the intercentriolar linker proteins, C-Nap1 and rootletin. 3 Ectopic overexpression of wild-type Nek2A in interphase cells induces premature splitting of centrosomes, providing a defined phenotype by which Nek2A activity in cells can be measured independently from other mitotic kinases. Here, we made specific use of a U2OS cell line with tetracycline-inducible Nek2A overexpression, U2OS:GFP-Nek2A, that had been generated previously in our laboratory. 11
Asynchronous U2OS:GFP-Nek2A cells were either uninduced or induced with doxycycline alone or together with DMSO, CC004731, or CC004733 as active viridin-like compounds or CC04730, a structurally related but inactive viridin-like compound. After 24 h, cells were fixed, stained for γ-tubulin to detect centrosomes, and analyzed by immunofluorescence microscopy (
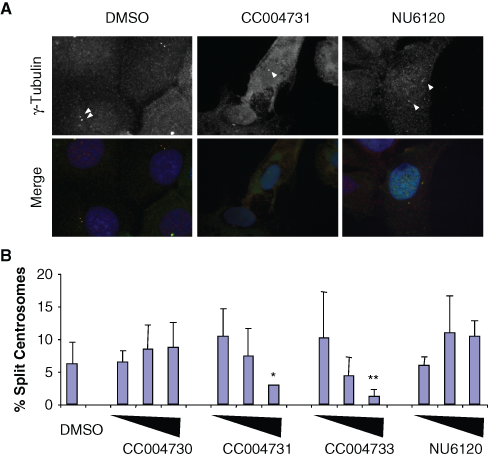
Inhibition of Nek2-induced centrosome splitting by viridin compounds. (
Automated phenotypic assay for Nek2 inhibition
To overcome the laborious process of scoring centrosome splitting manually, we automated the microscopy assay described above. To determine the best fluorescence marker of centrosome localization, we tested a number of antibodies. Centrosomes appeared relatively weak when stained with antibodies against γ-tubulin or C-Nap1 (data not shown). This observation was made under noninduced conditions and may be due to higher than normal levels of Nek2A expression, which may to lead to their displacement from the centrosome.
2
In contrast, a strong S/N ratio for centrosomes was obtained by staining with an antibody against the pericentriolar material marker pericentrin (
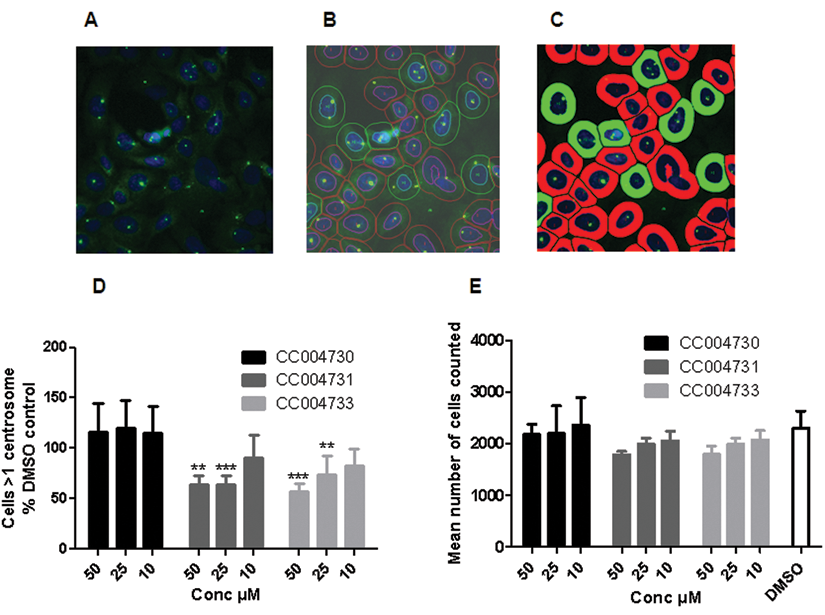
Automated cell-based assay for Nek2 inhibition. (
Discussion
Although cancer drug discovery programs are well advanced for several mitotic kinases (e.g., Cdk, Plk, and Aurora kinases), interest has only recently turned to the NIMA-related kinases. There is good evidence that several members of this family have mitotic roles, including Nek2, Nek6, Nek7, and Nek9. Expression and depletion studies to date support the notion that Nek2 could indeed be an important anticancer target. 3,7,8 Thus, we undertook an unbiased high-throughput screen to identify small-molecule inhibitors of Nek2 kinase activity.
Screening assays for Nek2 activity that use either a heterogeneous or homogeneous time-resolved fluorescence endpoint have been described. 18 We employed a radiometric proximity assay based on the use of solid scintillant-coated microplates as, when we commenced this work, Nek2 phosphorylation sites had not been identified and nonisotopic assays based on the use of generic serine/threonine kinase substrates and phosphospecific antibodies did not prove successful. The optimized radiometric assay included ATP concentrations below the Km as determined in a gel-based kinase assay and was robust and reproducible with batch variation of ~10% CV and overall Z′ factors >0.6. The assay was used to screen a compound library with highly acceptable overall performance. Several classes of hit structures were found, and these are among the first small-molecule inhibitors of Nek2 to be reported.
The hits included nonselective toxoflavin kinase inhibitors and analogs of the natural product viridin. Both the toxoflavins and molecules containing the viridin core are recognized as having the potential to chemically react with proteins. For toxoflavins and other 7-azapteridines, this often involves redox reactions with exposed thiol groups. 21,25,30 It is interesting to note that the X-ray structure of Nek2 shows 2 cysteine residues in the ATP-binding site. 31,32 The chemical reactivity of wortmannin with PI3 kinase has been characterized as reaction with Lys802 in the ATP-binding site through nucleophilic attack and ring opening of the strained furan ring. 26,33 The viridin analogs from this screen may undergo similar covalent binding to an appropriately positioned lysine. Indeed, the Hill slopes for the 2 most active compounds (CC004731 and CC004733) were relatively high (~1.5), whereas those determined for staurosporine approximated 1.0 (data not shown). This may support the notion that these particular compounds are chemically reactive. The size and calculated physicochemical parameters (MW = 207-350, ClogP = 0-3) of the toxoflavins and viridin analogs identified here are within the acceptable limits proposed by several groups for the development of targeted molecular therapeutics. However, the possibility of a nonspecific mechanism of action through chemical reactivity may increase the risk of off-target toxicities that could frustrate clinical use. The identification in our screen of other drug-like compounds originating from a kinase-focused library that do not contain potentially reactive groups demonstrates that more attractive starting points for drug development against Nek2 can be found.
Interestingly, the viridin-like compounds showed a high degree of specificity within the mitotic members of the Nek kinase family with more than 70-fold more activity against Nek2 than Nek6 or Nek7. On the other hand, these compounds generally exhibited similar levels of activity against Plk1, Aurora A, and Cdk1 as against Nek2. These data highlight the importance of having structural information on how inhibitors bind to the active sites of different kinases, as such information allows decisions to be made on chemical modifications that would improve selectivity. However, to date, only a limited number of high-resolution structures of the Nek2 kinase domain have been solved. The first used a construct containing an inactivating point mutation (T175A) in the T loop. 31 Moreover, this structure was obtained in the presence of the generic kinase inhibitor, SU11652. Further high-resolution structures have now been described using both the human wild-type and mutated (T175A) kinase domain of Nek2, 32 although it remains to be seen whether these will be representative of full-length Nek2 in the presence of small-molecule inhibitors, such as the viridins.
As predicted from Nek2 depletion studies, 7,8,13 2 of the active viridin compounds, but not the inactive analog, showed antiproliferative properties against 2 human cancer cell lines. However, because of the known promiscuity of this compound class, growth inhibition resulting from nonselective inhibition of other kinases cannot be ruled out, as illustrated by the effect of wortmannin on proliferation.
Importantly, the active viridin-like compounds, but not the closely related compound that lacked activity against Nek2, affected premature centrosome splitting in a human tumor cell line. Overexpression of Cdk2/cyclin A or Cdk2/cyclin E, but not Plk1, Aurora A, Cdk1/cyclin B, or Cdk4/cyclin D, can also induce significant levels of premature centrosome splitting. 29 However, the splitting induced by active Cdk2 occurs as the result of a separate pathway from that induced by Nek2A. 29 Furthermore, a compound, previously published as a Cdk2 inhibitor 28 but that lacked activity against Nek2, did not block centrosome splitting in the Nek2A-inducible cells. This is consistent with the active viridins acting as direct Nek2 inhibitors in cells.
In view of these promising results, we explored the possibility of automating the manual image-based assay for use as a secondary screen in an ongoing drug discovery project. Antibodies against the PCM marker, pericentrin, gave a single bright spot when centrosomes were unsplit but 2 distinct dots when centrosomes were split. By additional staining of nuclei and arbitrarily setting an approximate cell perimeter based on the nuclear area, we developed an algorithm that could count the number of pericentrin-stained centrosomes per cell. Automated counting of Nek2A-inducible cells treated with DMSO or the viridin-like compounds demonstrated that this assay was capable of scoring cellular inhibition of Nek2A and would potentially allow more rapid and efficient estimation of cellular potency (EC50) for compounds generated during medicinal chemistry optimization of hit compounds.
In summary, we have successfully completed a small-molecule high-throughput screen to identify chemical inhibitors of Nek2 kinase activity. Several series of compounds with activity against Nek2 kinase activity were identified. These included a series of toxoflavins and steroid-like, viridin compounds that demonstrated the validity of the screen. Results with the viridin hits show that pharmacological inhibition of Nek2 kinase results in the expected phenotype of disruption to centrosome function that is associated with growth inhibition and further support Nek2 as a target for cancer drug discovery.
Footnotes
Acknowledgements
We thank all members of our laboratories for useful discussion and are grateful to Prof. R. Griffin (Newcastle) for supply of the NU6120 compound and GE Healthcare for advice in setting up the cell-based assay.
This work was supported by grants to AMF, WA, and PW from the Discovery Committee of Cancer Research UK. The Cancer Research UK Centre for Cancer Therapeutics is funded primarily by Cancer Research UK program grants C309/A2187 and C309/A8274. We acknowledge NHS funding to support the work of the NIHR Biomedical Research Centre, the ICR, and The Royal Marsden NHS Foundation Trust.
The authors have no conflicts of interest to declare.