
Abstract
Huntington’s disease (HD) is a fatal neurodegenerative disease characterized by progressive cognitive, behavioral, and motor deficits and caused by expansion of a polyglutamine repeat in the Huntingtin protein (Htt). Despite its monogenic nature, HD pathogenesis includes obligatory non-cell-autonomous pathways involving both the cortex and the striatum, and therefore effective recapitulation of relevant HD disease pathways in cell lines and primary neuronal monocultures is intrinsically limited. To address this, the authors developed an automated high-content imaging screen in high-density primary cultures of cortical and striatal neurons together with supporting glial cells. Cortical and striatal neurons are transfected separately with different fluorescent protein markers such that image-based high-content analysis can be used to assay these neuronal populations separately but still supporting their intercellular interactions, including abundant synaptic interconnectivity. This assay was reduced to practice using transfection of a mutant N-terminal Htt domain and validated via a screen of ~400 selected small molecules. Both expected as well as novel candidate targets for HD emerged from this screen; of particular interest were target classes with close relative proximity to clinical testing. These findings suggest that composite primary cultures incorporating increased levels of biological complexity can be used for high-content imaging and “high-context” screening to represent molecular targets that otherwise may be operant only in the complex tissue environment found in vivo during disease pathogenesis.
Keywords
Introduction
H
This property of mutant Htt has enabled the development of a range of in vitro and in vivo assays as screening platforms for HD drug discovery. 10,11 These include a number of molecular assays that are designed to target aggregation of polyglutamine- containing peptides or proteins that have successfully identified compounds subsequently shown to also have anti- aggregative properties in cell and animal models (e.g., Apostol et al., 12 Desai et al., 13 Wang et al., 14 and Berthelier and Wetzel 15 ). More recent research, however, has fuelled debate over whether larger aggregates are inherently toxic, cytoprotective, or epiphenomenal 16 (reviewed in Truant et al. 17 ). Cell-based HD assays have generally focused on downstream targets of mutant Htt, such as surrogates of cell death (caspase-3 activation or lactate dehydrogenase release) as the assay endpoints (e.g., Aiken et al., 18 Piccioni et al., 19 Varma et al., 20 and Wang et al. 21 ). A number of compound hits from these screens have also been tested in more complex tissue or animal model systems such as Drosophila and transgenic HD mouse models. 9,22,23
However, the rate of success of these compounds has been relatively low, even in the context of de novo drug discovery, in terms of continued efficacy in increasingly physiologically and clinically relevant preclinical models, suggesting that key aspects of HD pathogenesis may not be adequately represented in conventional cell-free and cell-based HD models. Moreover, although striatal degeneration has long been a focus of HD research, increasing evidence suggests the complicit dysfunction of other brain regions, notably the cerebral cortex, and indeed neurodegeneration in late-stage HD is pervasive and widespread across the CNS. 24-27 As such, HD may not fundamentally be a cell-autonomous disorder but rather one that in vivo involves the interaction of several cell types, notably both striatal and cortical neurons. 24,25 In fact, basic aspects of striatal neuronal differentiation have been demonstrated to not occur in vitro in the absence of co-culturing with and active synaptic input from cortical neurons. 28
For new HD drug and drug target discovery, it would therefore be desirable to have an intermediate-stage screening assay with increased representation of the actual cell types and intercellular interactions that drive key aspects of HD pathogenesis in vivo, yet with sufficient throughput and assay robustness to support such applications as hit-to-lead, lead optimization, and primary screening of targeted compound libraries. In this context, we describe here a novel “composite” primary neuronal screening assay based on high-content analysis (HCA) that incorporates both striatal and cortical neurons in combination with their native glial cell counterparts. This approach offers significant advantages over cell line and primary cell monoculture assays in terms of physiological and clinical relevance, yet the use of image-based HCA allows the deconvolution of individual neuronal species within these composite cultures into cell-type-specific assay readouts even on the background of such a complex biological milieu. 29
Using this high-content, high-context approach, we screened an initial set of ~400 small-molecule compounds implicated in HD by their presumptive target mechanisms or by their origin from other cell-free or cell-based screens for HD. These compounds included 27 compounds with preexisting efficacy data in animal models of HD or related diseases/mechanisms. Of the hits emerging from this initial screen, we describe 4 compound target classes that provided significant and reproducible neuroprotection in this novel cortico-striatal primary neuronal assay and that are of particular interest because of the proximity of these drug/target classes to clinical use. Together, these results indicate the feasibility of automated high-content screening in the context of multiple, interconnected neuronal cell types in a complex co-culture system.
Materials and Methods
Plasmids
The HttN90Q8 and HttN90Q73 expression plasmids were constructed based on clones and sequences that were generous gifts of Dr. Chris Ross (Johns Hopkins) and the Hereditary Disease Foundation and contained Htt exon-1 (90 aa) with an expanded CAG repeat domain encoding 8 or 73 glutamines, respectively, cloned into the expression vector gWiz (Genlantis, San Diego, CA). Independent reporter constructs for yellow fluorescent protein (YFP), cyan fluorescent protein (CFP), and mCherry were also made in the gWiz backbone.
Primary culture and transfection
Striatal and cortical tissues from E18 embryonic brains were microdissected and dissociated enzymatically with papain/DNase I (Worthington Biochemical Corp., Lakewood, NJ). Then, 5 × 106 cells of each neuronal cell type were electroporated separately using an Amaxa device (Lonza, Basel, Switzerland) with striatal versus cortical neurons co-transfected with different fluorescent protein reporter constructs to allow subsequent individual tracking under HCA. After electroporation, striatal and cortical cells were mixed together and then plated into 96-well plates in which glial beds had been plated 3 days previously. These final mixed cultures of neurons and astrocytes were grown in Neurobasal medium (Invitrogen, Carlsbad, CA) supplemented with 5% fetal calf serum (Sigma-Aldrich, St. Louis, MO), 2 mM glutamine (Glutamax, Invitrogen), 10 mM potassium chloride, and 5 µg/mL gentamicin.
Astroglia were generated from excess cortical cell preparations during the neuronal dissection by plating the mixed cell preparation into T-75 flasks (Becton Dickinson, Franklin Lakes, NJ) in Neurobasal medium (see above) (P0). The P0 cell mixtures were passaged into new flasks (P1) after 4 days in vitro (DIV). Thereafter, astroglial preparations were given media changes after 3 days and passaged after 7 days (>90% confluence). After 2 passages (P2), most of the nonglial cells had been removed, and cells were passaged into T-150 flasks for the third passage (P3). Astroglia were used for experiments at the P3 or P4 passage. For plating into 96-well plates, glia were harvested with trypsin/EDTA (Invitrogen), counted, and seeded at a density of 2000 cells/well into poly-D-lysine-coated 96-well plates (Becton Dickinson). These glial beds were used 3 to 4 days later for neuronal plating (see above).
Immunostaining and confocal microscopy
For immunostaining, identical cell mixtures were plated at equivalent densities on poly-lysine/laminin-coated coverslips (Becton Dickinson) in 24-well plates previously seeded with glial feeder layers as described above. When plating into 24-well plates, glial and neuronal cell numbers were scaled to equivalent cell densities as in the 96-well plate cultures. To immunostain, medium was removed and cells were fixed in freshly diluted 4% paraformaldehyde (16% stock, VWR)/4% sucrose for 20 min and then rinsed twice with phosphate- buffered saline (PBS). Cells were blocked with 5% goat serum/2% bovine serum albumin (BSA)/0.1% Triton X-100 in PBS for 1 h at room temperature, followed by incubation with primary antibodies overnight at 4°C in a humidified chamber. Coverslips were washed 4 times for 5 min each in PBS and appropriate secondary antibodies applied at 1:500 (4 µg/mL) in blocking solution for 2 h in the dark followed by 4 rinses with PBS. Finally, coverslips were mounted in Vectashield (Vector Labs, Servion, Switzerland) for imaging. Antibodies were purchased from Millipore (Billerica, MA) or Sigma-Aldrich (St. Louis, MO) and used at the following dilutions: NeuN 1:500, DARPP-32 1:100, vGLUT1 1:1000, GAD67 1:1000, synaptophysin 1:100, and spinophilin 1:400.
Electrophysiology
For electrophysiological recording, cortico-striatal co- cultures were prepared as described above for immunostaining. Portions of coverslips were then transferred on the specific DIV to a recording chamber, where standard patch-clamp methods 30 were used to obtain whole-cell recordings from striatal neurons identified by their fluorescence labeling. Briefly, patch-clamp electrodes (~3 MΩ resistance) were used to record synaptic currents on an Axopatch 200B voltage clamp with pClamp version 9 software (Molecular Devices, Sunnyvale, CA). The neurons were held at −65 mV, and signals were digitized at 5 kHz followed by low-pass Bessel filtering at 2 kHz. For recording miniature excitatory postsynaptic currents (mEPSCs), the extracellular solution contained (in mM) the following: 137 NaCl, 3 KCl, 2 CaCl2, 10 glucose, and 10 mM HEPES, adjusted to pH 7.4 with NaOH. Then, 10 µM tetrodotoxin (TTX) and 20 µM bicuculline were added to block voltage-gated sodium channel and GABA receptor currents, respectively. The internal recording solution contained (in mM): 130 CsCl, 0.2 EGTA, 4 MgATP, 0.3 Na2GTP, and 10 mM HEPES, adjusted to pH 7.3 with CsOH. To record miniature inhibitory postsynaptic currents (mIPSCs), the same solutions were used, but the bicuculline was replaced with 10 µM NBQX and 10 µM APV to inhibit AMPA and NMDA currents. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. Offline data analysis for mEPSC and mIPSC activity was done using Clampfit 9 (Molecular Devices) and mini analysis software from Synaptosoft (Decatur, GA).
High-content screening and analysis
Compound screening was done in a 96-well plate format with striatal and cortical neurons transfected separately with HttN90Q73 in combination with YFP, CFP, or mCherry, n = 6-8 wells for each condition. Positive control wells were treated with 10 µM KW-6002 or transfected with YFP and gWiz-CFP as a non-Htt transfection control. Negative control wells were treated with 0.5% DMSO, the carrier for compound delivery. Images of cells were captured on the Cellomics Arrayscan VTI (Thermo Scientific, Pittsburgh, PA) using a 10×/0.3 NA objective through appropriate filter sets for YFP, CFP, or dsRED (Chroma Technology, Rockingham, VT) and analyzed using the Cellomics Target Activation algorithm. Initially, 16 fields per well were acquired at full resolution; however, after statistical determination (data not shown), this was reduced to 9 fields per well. The algorithm was optimized for object size, object shape, and fluorescence intensity to identify specific neuronal cell bodies, as further described in the main text. Data graphs were generated, and statistical significance and nonlinear regression were calculated using GraphPad Prism (GraphPad Software, La Jolla, CA). Statistical significance was determined by 1-way analysis of variance (ANOVA) followed by Dunnett’s post hoc comparison test at the 0.05 confidence level. The z′ factor was calculated from a consecutive 60-plate run in a 1-month period. 31
Test compounds
Test compounds were acquired through the CHDI Foundation, Inc. (Los Angeles, CA) or purchased directly from Tocris Bioscience (Ellisville, MO), Sigma-Aldrich (St. Louis, MO), or Calbiochem/EMD Biosciences (Gibbstown, NJ). Compounds were dissolved in 100% DMSO and diluted in culture medium to 2× the desired final concentrations, then added 1:1 (vol:vol) to culture wells immediately after neuronal plating. Final concentration of DMSO in the assay was 0.5% for all assay wells unless otherwise noted. Brain-derived neurotrophic factor (BDNF) (Sigma-Aldrich) was reconstituted in PBS and stored at −70°C in single-use aliquots and diluted for use in a similar fashion except for no addition of DMSO.
Results
Assembly of composite cortico-striatal cultures
The goal of these studies was to develop a high-content, high-context primary neuronal screening assay in which specific neuronal cell types such as striatal neurons and cortical neurons could be tracked as distinct cell populations while remaining resident—and synaptically interconnected—in a complex, multicellular mixture, including multiple neuronal and glial cell types. In diseases such as HD, in which intercellular interactions among different neuronal and glial populations appear to mediate core components of disease pathogenesis, such a screening assay could be particularly important in capturing non-cell-autonomous processes critical for disease pathogenesis.
We reasoned that such a screening assay could be constructed by combining automated HCA with high-density mixed primary cultures of neurons and glia from both striatum and cortex, the 2 major brain areas implicated in HD initiation and progression, providing that striatal and cortical neurons could be selectively labeled with different and independent fluorochromes for tracking distinct cell types and cell populations within these mixed cultures. To accomplish this, we microdissected striatal versus cortical brain tissues from E18 rat embryos and then dissociated and electroporated these different neuronal populations separately with different wavelength fluorescent protein-encoding indicator plasmids. These neuronal populations were then recombined and plated onto previously established glial cell support layers in 96-well plates (
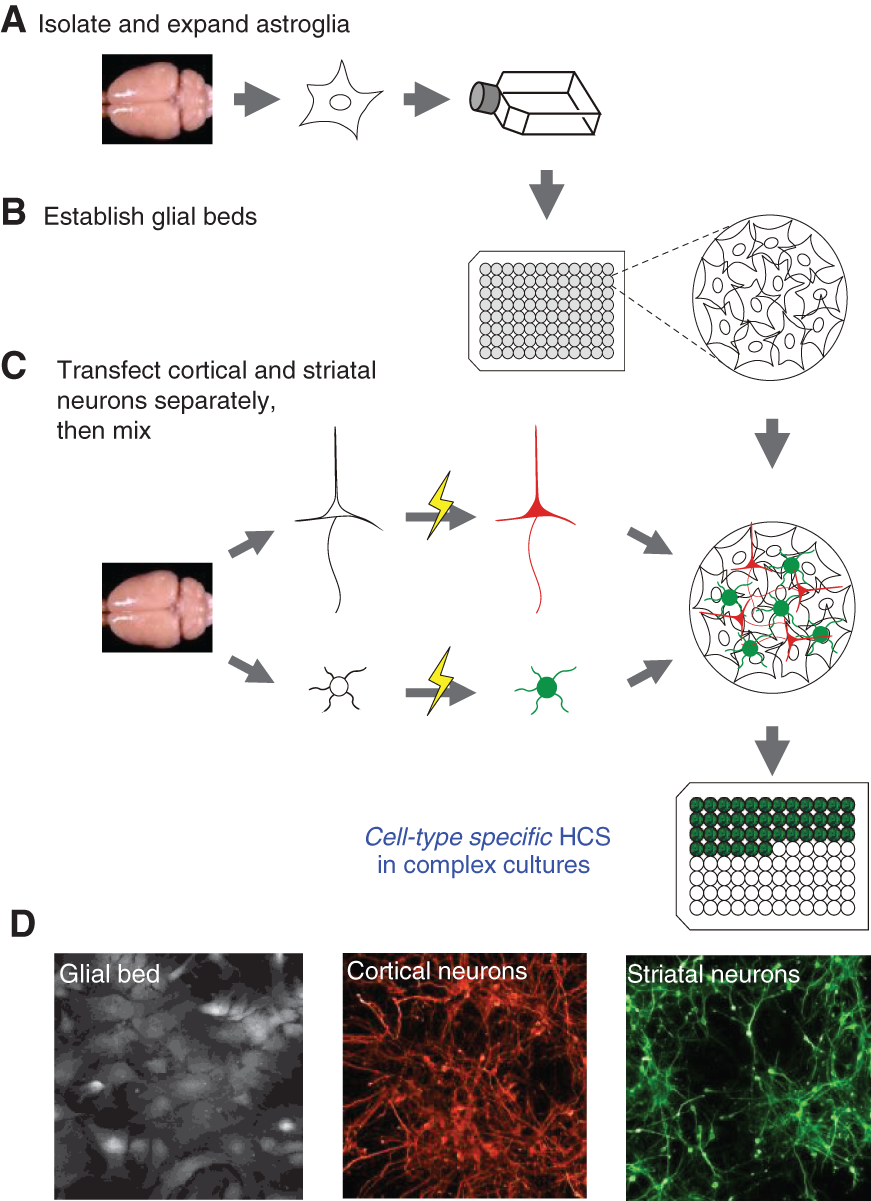
Assembly process for composite cortico-striatal co-cultures. (
Such mature glial beds are known to be critical for neuronal survival and development in vitro and are supportive of the reestablishment of synaptic connectivity between cortical and striatal neurons as is the aim of this assay strategy. Glial cells were prepared from isochronic rat brain tissues and expanded by 3 passages in culture before seeding into 96-well plates 2 to 3 days before neuronal dissection to allow for prior establishment and maturation of glial beds (
In fact, the cell populations expressing fluorescent reporters were of almost exclusive neuronal morphology (
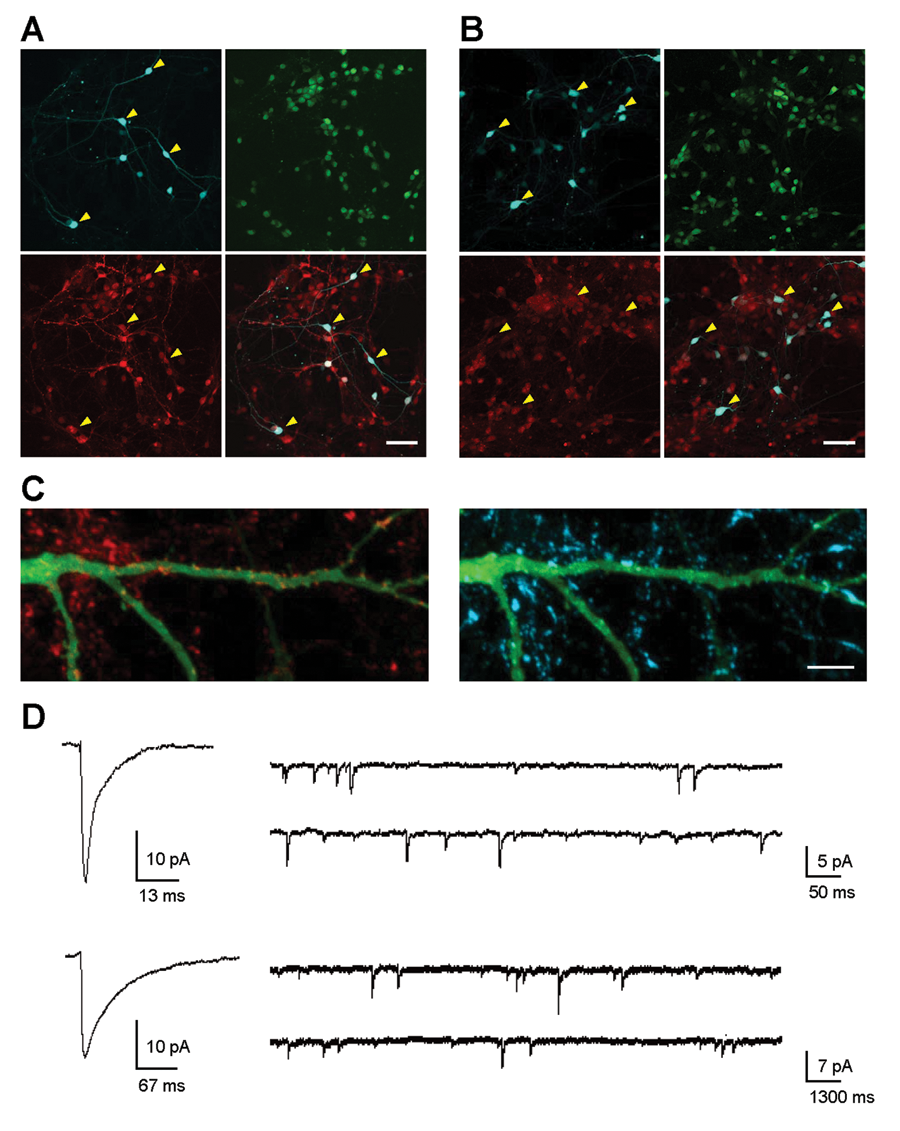
Striatal and cortical neurons in composite cultures express cell-type-specific markers and are electrically and synaptically active. (
Finally, we used whole-cell patch-clamp recording to determine directly if co-cultured striatal and cortical neurons were electrically active and had reestablished the expected types of synaptic connections (
High-content analysis of striatal versus cortical neurons in co-cultures
We next developed and optimized automated high-content microscopy imaging algorithms on the Cellomics ArrayScan VTI platform (ThermoFisher, Pittsburgh, PA) to identify and analyze the comingled but differentially labeled striatal versus cortical neurons residing in these composite cultures in a 96-well format. Fluorescent proteins (CFP, YFP, or mCherry) were excited with appropriate excitation filters using a metal halide light source (EXFO, Ontario, Canada). Fluorescence emissions were collected using CFP, YFP, or DsRed filter sets (Chroma Technology, Rockingham, VT) through a 10×/0.3 NA objective and images captured with a 12-bit Orca-II ER CCD camera (Hamamatsu, Hamamatsu, Japan) to positively identify fluorescent objects based on a defined threshold above background in the field using the Cellomics Target Activation protocol. Either 9 or 16 image fields per well were collected at full resolution (1024 × 1024 pixels) with an average fixed exposure time ranging from 0.05 to 0.5 s per fluorescent channel. The algorithm used to identify neuronal cell bodies was optimized based on both morphology and intensity properties of objects, and a segmentation algorithm was employed to separate objects in close proximity; large clusters or clumps of cells, which were rare, were excluded from analysis. As indicated above, relatively few transfected astroglia cells were observed during image analysis and were easily discriminated by their large object size and shape compared to either striatal or cortical neurons (
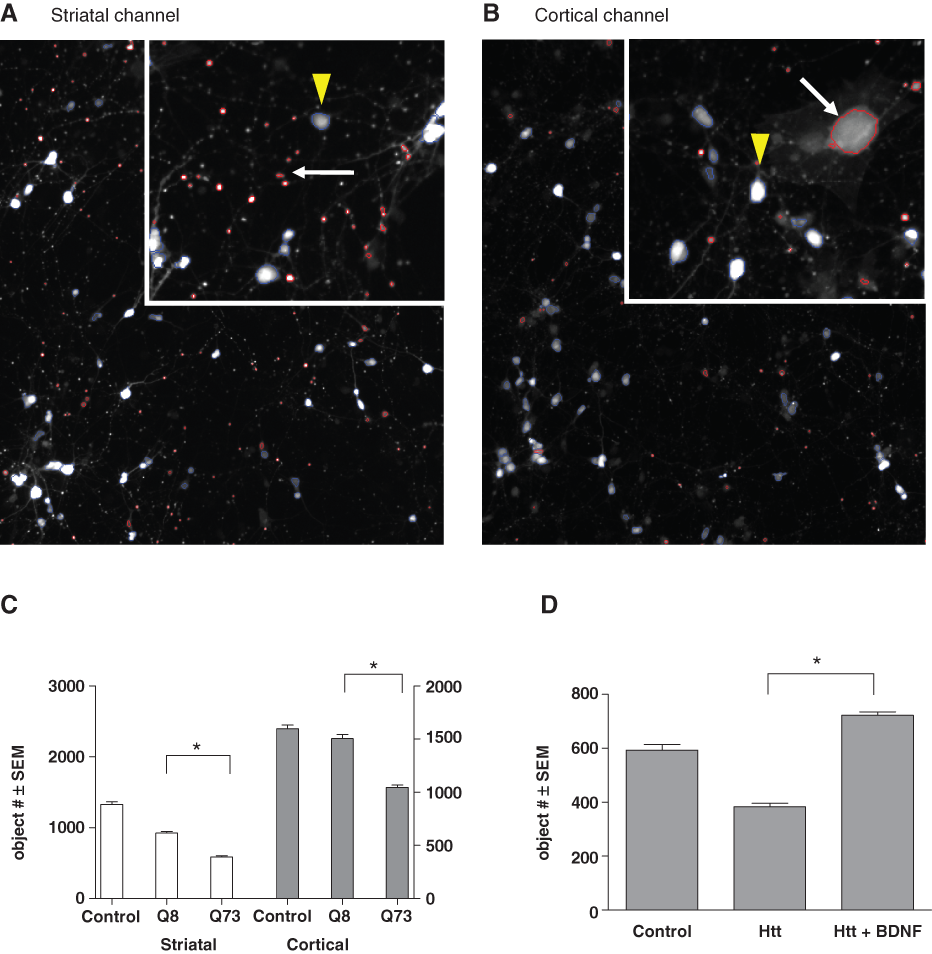
High-content analysis (HCA) of neurons coexpressing fluorescent proteins and an N-terminal domain of mutant Htt. Cellomics ArrayScan VTI generated images showing striatal neurons expressing (
To model HD in these composite cultures, we cotransfected an Htt exon-1 sequence carrying a repetitive CAG sequence encoding a73 polyglutamine expansion (“HttN90Q73”), the same Htt fragment with an 8 CAG repeat corresponding to the endogenous rat gene (“HttN90Q8”), or a fluorescent protein plasmid (CFP) together with either YFP (into striatal neurons) or mCherry (into cortical neurons), each hereafter referred to as the “control” condition for these 2 neuronal populations, respectively. As the cotransfection linkage rate was relatively high at ~80% (Suppl. Fig. S1), we could expect that the majority of striatal and cortical neurons that expressed a fluorescent reporter also expressed the cotransfected Htt construct.
Transfection of polyglutamine-expanded HttN90Q73 induced the progressive degeneration of both striatal and cortical neurons over the course of 6 DIV or more. At 2 DIV, no differences were seen between control and HttN90Q73-transfected cultures, but by 4 to 6 DIV, there were significantly fewer neurons of either species surviving in HttN90Q73-transfected cultures (
Thus, to quantify the potential neuroprotective effects of drug candidate compounds in this assay, cell object numbers were primarily analyzed at 6 DIV, at which the largest differences between control and HttN90Q73-transfected neuronal numbers were observed without necessitating media changes or refeeding of compounds. As an initial cross-validation of this assay relative to other, previously established HD models, we tested the ability of BDNF to protect striatal neurons against HttN90Q73-induced neurodegeneration in these composite cultures (
Compound screening using composite cortico-striatal cultures
For compound screening, the final 96-well assay format excluded the perimeter wells due to edge effects from evaporation and used n = 6 wells for each experimental condition. In this format, 7 compound concentrations could be evaluated per 96-well plate, with 3 conditions assigned to in-plate positive and negative controls. Data acceptance criteria required >100 fluorescent cells per control well and a minimum of 4 acceptable wells per control condition. In addition, a compound screening run was considered acceptable if the following criteria were met: >1.5-fold difference between the control and HttN90Q73 conditions and a statistically significant difference with p < 0.05 between HttN90Q73 and the positive control compound condition. Compounds were considered hits if the run met the above criteria and the compound showed p < 0.05 statistical significance compared to the HttN90Q73 condition by Dunnett’s post hoc comparison test following ANOVA.
Given an average of 12 E18 rat embryos per pregnant dam and a final plating density of 60,000 striatal and 60,000 cortical neurons per well, up to 8 such formatted 96-well plates could be prepared and run per rat litter. Using these parameters, the theoretical throughput for 4 runs/litters per week is 832 compounds/year at 7-point dose-response or 5824 compounds/year at single-point dose, depending on the level of statistical discrimination power desired.
Each run includes a positive control condition not transfected with Htt, as well as an HttN90Q73-transfected condition treated with a positive control compound. The z′ factor for the assay, 31 calculated from 60 compound plates run over a 1-month screening period, was determined to be 0.54 with an assay window (i.e., dynamic range) of 1.9, suggesting suitability for such screening uses of the assay.
In the initial implementation of this assay in screening mode, we evaluated a set of small-molecule compounds at 3- or 7-point dose responses under fully automated HCA. These compounds were collected as part of a research collaboration with the CHDI Foundation and were chosen on the basis of their nominal targets being implicated in HD by their identification in previous target- or cell-based HD screens. Using the criteria described above, the hit rate for this initial screen was 8%, with 31 compounds showing significant neuroprotection (Suppl. Table S1), whereas 37 compounds exhibited cytotoxicity in the co-culture assay (data not shown).
Among the compound screening set were 28 compounds representing clinical candidates from the Huntington Study Group (http://www.huntington-study-group.org/) and/or have been evaluated in animal models of HD (Suppl. Table S2). Of these, 7 compounds provided significant levels of neuroprotection to HttN90Q73-transfected striatal neuron in co-culture, representing a range of different target classes, including heat shock proteins, serotonin modulation, neurotrophin signaling, and energy metabolism. Other included compounds, such as tetrabenazine, although clinically effective, have not been reported to provide direct neuroprotective support and did not show neuroprotection in cortico-striatal co-cultures. Interestingly, antiapoptotic compounds, including BOC-D-FMK and HDAC inhibitors, generally failed to show significant neuroprotection in this assay. Together, these results suggested that the composite cortico-striatal co-culture assay described here has the potential to identify neuroprotective compounds that may be predictive of continued effectiveness when tested in subsequent whole-animal model studies.
Of the remaining hits, we describe 4 target classes here that supported neuroprotective activity for multiple compounds with dissimilar chemical structures:
Rho kinase (ROCK) inhibitors
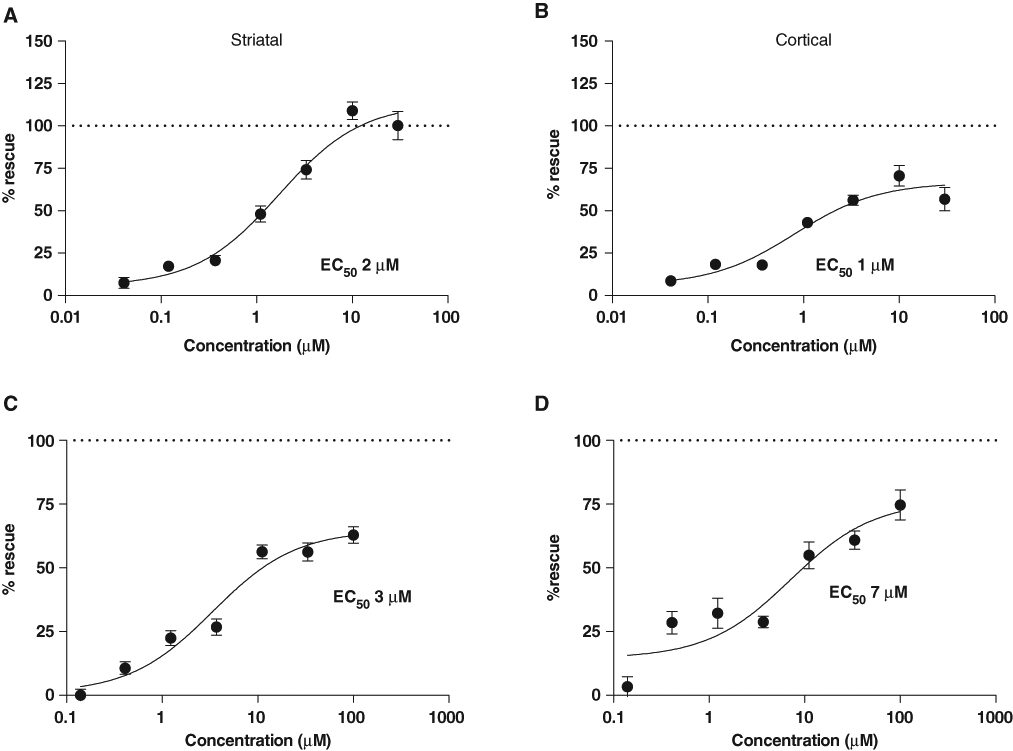
The ROCK inhibitory compound ROCK2_Ex_55 showed clear neuroprotection for both striatal and cortical neurons transfected with HttN90Q73 in composite co-cultures, with EC50s of 2 µM and 3 µM, respectively (
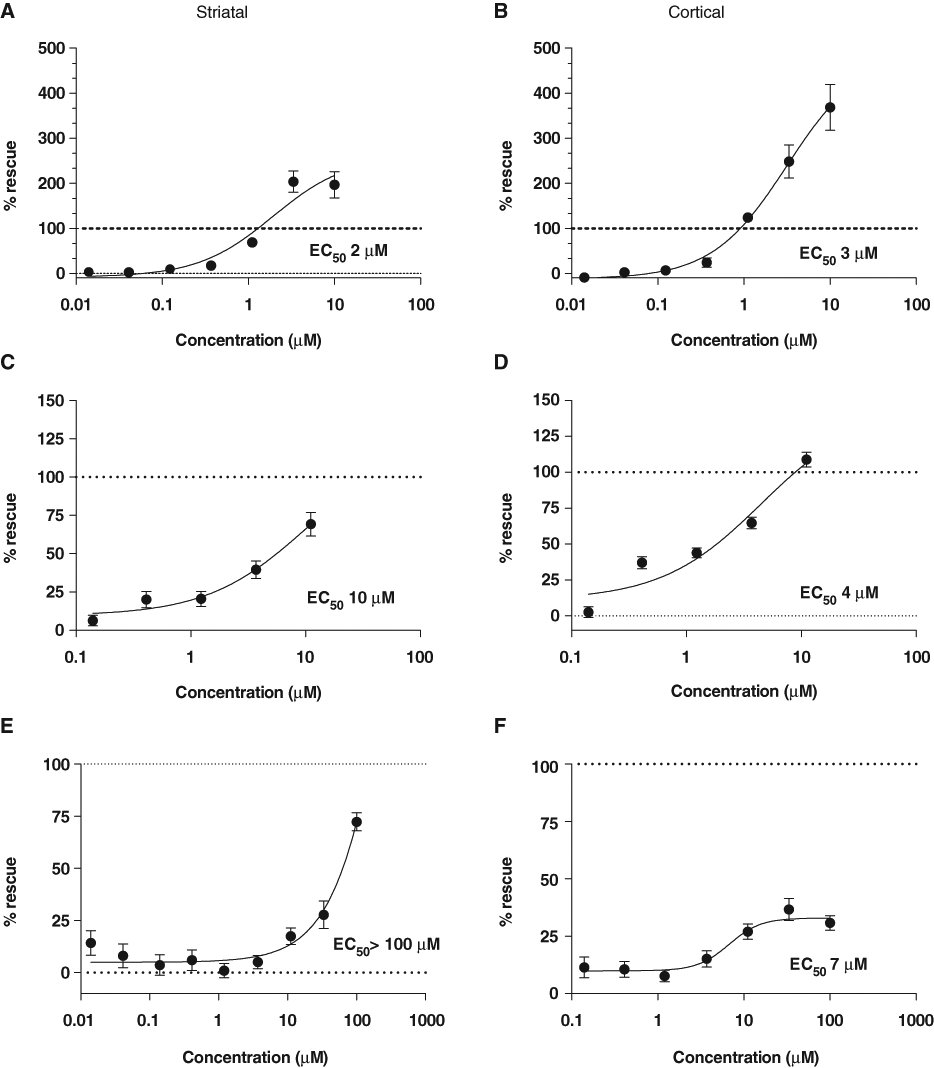
Rho kinase (ROCK) inhibitors are protective against HttN90Q73-induced neurodegeneration. Neuroprotective activities of these ROCK inhibitors were identified as part of a targeted screen of ~400 small-molecule compounds implicated in Huntington’s disease (HD), conducted as described in the main text and methods. (
Y-27632 was initially identified in a cell-based assay for polyglutamine aggregation inhibitors 34 and was later shown to improve motor function in the R6/2 mouse model. 35 In addition, Rho kinase inhibitors are in clinical development for cardiovascular disease and glaucoma and in preclinical development for a number of other disease states. 36,37 However, we were unable to detect neuroprotective effects of the Food and Drug Administration (FDA)–approved rho-kinase inhibitor fasudil (HA-1077) for concentrations up to 100 µM or for its active metabolite hydroxyfasudil (data not shown). Given known issues with in vitro target specificity of ROCK inhibitors, 38 it cannot be excluded that Y-27632 and Y-39983 may have additional kinase targets. 39
Phosphodiesterase (PDE) inhibitors
We identified 3 PDE4 inhibitors, including roflumilast and, to a lesser degree, rolipram, to be neuroprotective for HttN90Q73-transfected neurons in co-culture (
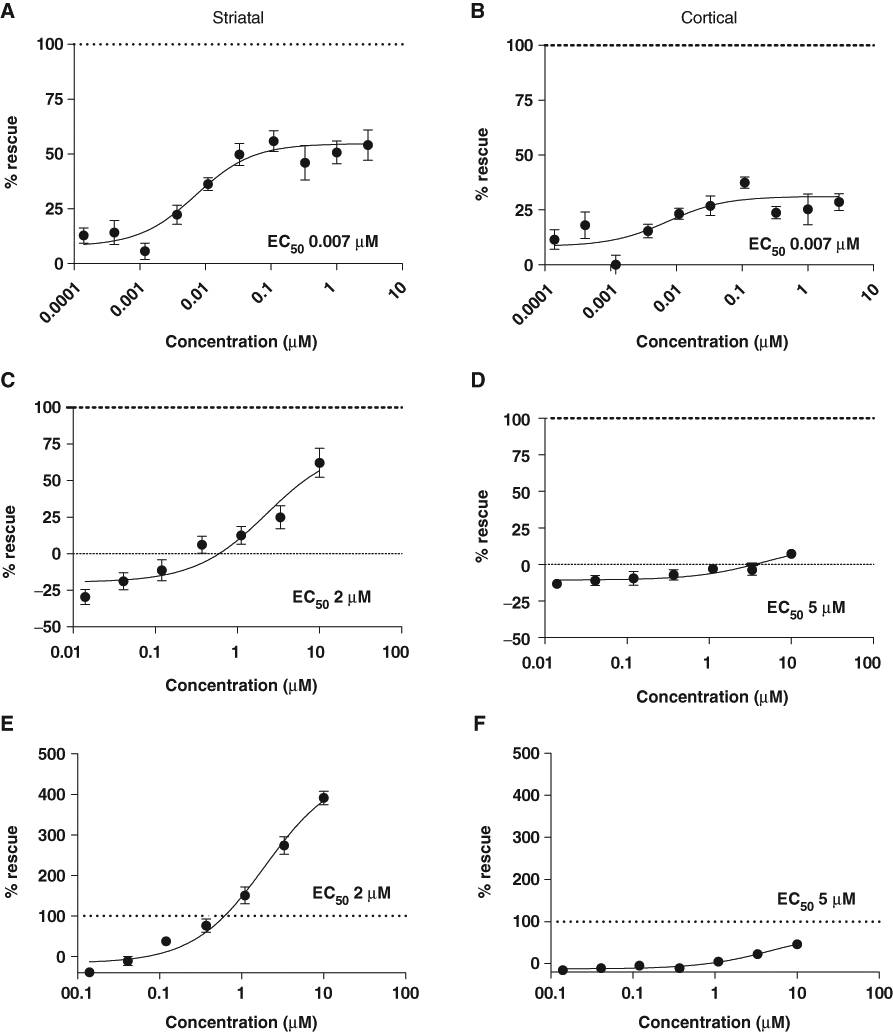
Compounds that alter cAMP signaling are neuroprotective against HttN90Q73-induced striatal and cortical neurodegeneration. Neuroprotective activities of cAMP modulators were also identified in the course of the targeted screen of ~400 small-molecule compounds as described in the main text and methods. (
Adenosine 2a receptor and IKKβ inhibitors
In addition, of note were 2 isolated hits that provided strong neuroprotection but were not as clearly corroborated by initial surveys of other compounds directed at the same nominal targets. The first was the adenosine 2a receptor antagonist KW-6002 (istradefylline), which provided robust and reproducible neuroprotection to HttN90Q73-transfected striatal neurons with an EC50 of 2 µM (
A2a receptor and IKKβ modulators. (
Finally, MLN-1145, an IKKβ inhibitor, showed strong protection against HttN90Q73-induced neurodegeneration for both striatal and cortical neurons in co-culture with EC50s of 3 and 7 µM, respectively (
Discussion
We have developed and implemented a complex, multicellular neuronal screening assay to better reflect the biological complexity of HD in the early stages of the drug discovery process. In particular, this assay seeks to capture non- cell-autonomous aspects of HD pathogenesis that involve the interaction of different cells types in different brain regions, including synaptic transmission. Thus, a key element of this assay is the co-culture of comingled striatal and cortical neurons transfected with a mutant Htt-based construct but in a process that allows each of these neuronal populations to be labeled independently with a different fluorescent marker. We demonstrate that image-based HCA can then be used to create phenotypic assays that are still restricted to individual, defined cell populations even within these densely mixed co-cultures. The initial screen of ~400 selected compounds encountered hits that validated this assay against compounds and targets already known to be effective in other models of HD and also provided direct evidence for new compound/target classes providing robust neuroprotection to striatal and/or cortical neurons in this system.
Together, these findings suggest that highly biological complex primary cell assays can be developed in which HCA can be used to develop rigorous, cell-type-specific phenotypic assays and thereby address important aspects of disease biology that may not otherwise be representable in conventional cell culture or other in vitro models. Important future developments will include the addition of other cell types that may be important to disease progression, such as microglia; the construction of additional functional endpoints directly addressing cell biological processes important to neuronal health and function, including dendrite outgrowth/regulation, calcium handling, electrical excitability, and synapse regulation and function; and the incorporation of RNAi methods for direct target validation studies.
Footnotes
Acknowledgements
We thank Vahri Beaumont and Gary Banker for helpful discussions on the design and implementation of the screen and Lan Ye for help with assay development.
This work was supported by the CHDI Foundation, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.