
Abstract
Techniques to evaluate gene expression profiling, including real-time quantitative PCR, TaqMan® low-density arrays, and sufficiently sensitive cDNA microarrays, are efficient methods for monitoring human embryonic stem cell (hESC) cultures. However, most of these high-throughput tests have a limited use due to high cost, extended turnaround time, and the involvement of highly specialized technical expertise. Hence, there is a paucity of rapid, cost-effective, robust, yet sensitive methods for routine screening of hESCs. A critical requirement in hESC cultures is to maintain a uniform undifferentiated state and to determine their differentiation capacity by showing the expression of gene markers representing all germ layers, including ecto-, meso-, and endoderm. To quantify the modulation of gene expression in hESCs during their propagation, expansion, and differentiation via embryoid body (EB) formation, the authors developed a simple, rapid, inexpensive, and definitive multimarker, semiquantitative multiplex RT-PCR (mxPCR) platform technology. Among the 15 gene primers tested, 4 were pluripotent markers comprising set 1, and 3 lineage-specific markers from each ecto-, meso-, and endoderm layers were combined as sets 2 to 4, respectively. The authors found that these 4 sets were not only effective in determining the relative differentiation in hESCs, but were easily reproducible. In this study, they used the HUES-7 cell line to standardize the technique, which was subsequently validated with HUES-9, NTERA-2, and mouse embryonic fibroblast cells. This single-reaction mxPCR assay was flexible and, by selecting appropriate reporter genes, can be designed for characterization of different hESC lines during routine maintenance and directed differentiation.
Keywords
Introduction
P
Reports from independent researchers have proposed that the hESC lines are somewhat similar in the expression of certain molecular markers implicating “stemness.” 15 Although not all markers have been tested for all cell lines, expression of a set of markers associated with pluripotency and differentiation capacity is accepted as a standard for most hESC lines. However, it is obvious that there are some subtle differences in the gene expression profiles between individual hESC lines. 9,10 This is not surprising given that each hESC line has been derived from a separate embryo, representing a unique genetic history. Furthermore, variations in the culture conditions followed by individual laboratories may produce certain genomic and epigenomic changes over time. These lead to difficulty in comparing data that are generated from one laboratory with that of another and sometimes even between different passages of the same cell line. It is undeniable that routine characterization of hESC lines is crucial to avoid compromising the integrity of results. One of the most common and effective methods of characterizing pluripotent stem cells is through reverse transcriptase (RT-PCR) using novel stage-specific genes that distinguish between hESC and EB. 16,17 Furthermore, the number of genes that can be considered reliable molecular markers for undifferentiated hESCs is relatively concise and also well documented. 18 Differentiated cells, on the other hand, can be identified by a number of lineage- and tissue-specific gene markers.
Previously, Chamberlain and coworkers 19 had demonstrated that PCR could simultaneously amplify multiple loci in the human dystrophin gene. Since then, multiplex PCR (mxPCR) has been well established as a routine technique for pathogen identification, gender screening, linkage analysis, forensic studies, template quantitation, and genetic disease diagnosis. mxPCR can be a 2-amplicon system or can also amplify separate regions of the same DNA. It may be the endpoint of analysis or preliminary to further analyses such as sequencing, hybridization, or real-time PCR depending on the objective and importance of the study. However, the steps for developing the mxPCR assay and the advantages of having multiple fragments amplified simultaneously are similar in either case.
Earlier we had proposed a characterization scheme of hESC, including RT-PCR, immunochemistry, karyotype, human leukocyte antigen (HLA) and short tandem repeat (STR) analyses, telomerase assay, teratoma formation in severe combined immunodeficient (SCID) mice, real-time PCR, and focused cDNA microarray, miRNA, and mitochondrial DNA analysis. 20,21 The objective of the present study was to characterize hESCs on a molecular level and to determine the quality and degree of variability among pluripotent cells and their early progenies (EB). Here, we illustrate an approach, based on single-reaction multiplex RT-PCR, for semiquantitative evaluation of hESC proliferation during ex vivo expansion. By comparing the relative mRNA levels of a set of carefully selected 15 markers in the hESC and EB samples, we could successfully discriminate between undifferentiated hESCs and their differentiated derivatives. We standardized the method using the HUES-7 cell line and further validated the technique not only with another independent hESC line, HUES-9, but also with a human teratocarcinoma cell line (NTERA-2) and MEF cells. Our data demonstrate that the technique developed here is simple, rapid, robust, and generally applicable for all cell lines. Hence, the combination of RT-PCR and related hESC-based technologies may provide a useful tool for setting a standard for hESC characterization in a cost-effective manner.
Materials and Methods
Propagation of hESC lines
HUES-7 and 9 were grown routinely on mitomycin C–inactivated (10 µg/mL; Sigma, St. Louis, MO) MEF feeder layers as described in our previous report. 21 Manual passaging was preferred over the enzymatic method to guarantee the best quality of hESCs for downstream characterization. Briefly, manual passaging was performed by mechanical dissociation of undifferentiated hESC colonies into small clumps of about 100 to 200 cells using the sharp edge of a flame-pulled Pasteur pipette under the stereomicroscope (Olympus; SZX16). We selectively picked the undifferentiated hESC colonies that were identified by morphological features, including large compacted cells with a higher nucleus-to-cytoplasm ratio and shiny borders. During every passage, utmost caution was adopted to ensure exclusion of spontaneously differentiated portions of the hESC colonies distinguished by their loosened distribution of relatively darkened cells lacking shiny borders and prominent nucleoli. Media were replenished every day, and passaging was done on the fourth to fifth days in culture. The cultures were maintained at 37°C and 5% CO2 in air (Binder, Camarillo, CA). For the NTERA-2 cell line, we used the same media as mentioned in our earlier report. 22
Generation of EBs in high-density suspension culture
For EB formation, hESC colonies were again manually dissected into small pieces of approximately 100 to 150 cells, which were then transferred onto ultra-low-attachment bacteriological dishes (Nunc, Rochester, NY) in media consisting of 80% Dulbecco’s modified Eagle’s medium (DMEM)/F-12 (GIBCO, Carlsbad, CA), 15% ES-tested fetal bovine serum (FBS; Hyclone, Logan, UT), 5% serum replacement (GIBCO), 1% nonessential amino acid solution (GIBCO), 1 mM glutamine (GIBCO), and 0.1% β-mercaptoethanol (Sigma). Undifferentiated hESCs spontaneously form EBs in suspension starting from day 2, implicating the onset of differentiation. Likewise, media were replaced every alternate day until the EBs had grown in size and maturity for up to 14 to 15 days.
Indirect immunofluorescence
Before immunostaining, hESC colonies were plated on MEF feeders in 2-well chamber slides (Nunc). Immunocytochemical analysis was performed as described in our earlier report. 3 Here, we have tested for undifferentiated stem cell and lineage-specific markers, including Oct-4 (Abcam, Cambridge, MA; ab19857), nanog (Abcam; ab62734), SSEA-4 (Millipore, Billerica, MA; MAB4304), TRA-1-61 (Millipore; MAB4360), GATA-4 (Abcam; ab61767), and nestin, α-SMA, and AFP (all 3 from Millipore; Human Embryonic Germ Layer Characterization Kit, SCR030). Cells fixed in 4% paraformaldehyde, stained against appropriate antibody coupled with a suitable conjugate (FITC or rhodamine), were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 10 µg/mL; Sigma, D9542), mounted, and examined under an Olympus microscope (BX51TR). Negative controls without primary antibodies were used for each marker in the study.
Total RNA isolation and cDNA synthesis
Test samples included hESC lines HUES-7 and 9, NTERA-2, and MEF cells. Cell pellets were collected and total RNA was isolated by the Trizol method (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. After estimation of the RNA (Nanodrop, Agilent, Santa Clara, CA), 1 µg of RNA treated with RNase-OUT ribonuclease inhibitor (Invitrogen) was used for cDNA synthesis. Broadly, 40 to 50 medium-sized hESC colonies yielded approximately 3 to 4 µg of total RNA. Reverse transcription using Superscript reverse transcriptase-II (Invitrogen) and Oligo dT (Invitrogen) to prime the reaction was carried out in 20 µL of reaction mix.
Gene-specific primers and uniplex and multiplex PCR
Combining the primers in various mixtures and amplifying several genes simultaneously ( Table 1 ) required alteration/optimization of some of the parameters of the reaction. When the multiplex reaction is performed for the first time, ideally equimolar amounts of primers are added similar to uniplex PCR. The subsequent results suggest how the individual primer concentration and other parameters need to be changed. All of the gene primers that we used in this study were already optimized using hESCs and their derivatives. 3 On the basis of our previous experience with routine molecular characterization of hESCs, gene-specific PCR primers were selected to distinguish between cDNA and genomic DNA. Then, 1 µL of cDNA was amplified by PCR using the Abgene 2× PCR master mix (Abgene, Epsom, UK) and appropriate primers ( Table 1 ) at concentrations ranging between 2 to 10 mm. PCR reaction was performed in 0.2-mL Eppendorf tubes (Axygen, Union City, CA) with a final volume to 12.5 µL. For all genes, amplification was performed for 35 cycles, consisting of an initial denaturation at 94°C for 5 min, then 94°C for 30 s; annealing temperature of the respective gene primer for 45 s, 72°C for 45 s; and termination by final extension at 72°C for 10 min and finally soaking at 4°C. All of the selected gene markers (see Table 2 ) were tested for amplification specificity and reproducibility at annealing temperatures ranging from 54°C to 60°C for optimization of the multiplex PCR assay platform. PCR products were analyzed on 2% agarose gel stained with ethidium bromide (Sigma), viewed under a UV trans-illuminator, and photographed on a Gel documentation system (Pharmacia Biotech, Piscataway, NJ). A 100-bp ladder (Invitrogen and Fermentas, Glen Burnie, MD) was used as molecular weight markers.
List of Gene Primers Classified in Separate Sets Along with the Forward and Reverse Sequences, Annealing Conditions, and Region of Amplification by the mxPCR Technique
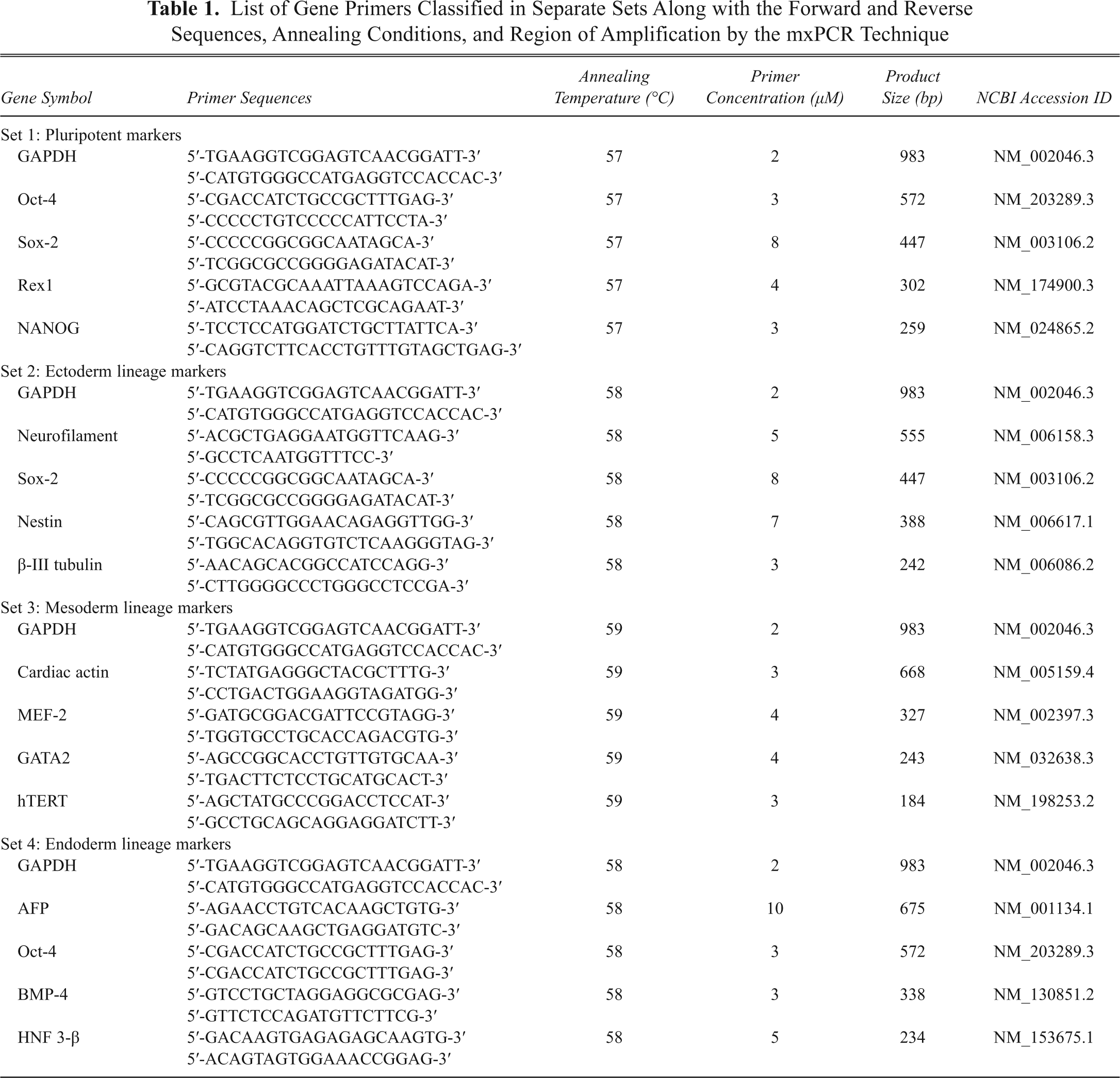
Details of the Gene Markers Used in This Study

Basic principles of the multiplex PCR
Based on numerous experiments, a standard protocol for multiplex PCR for characterization of hESC has been designed, including a number of practical solutions to some of the most commonly encountered problems. The list of various factors that can influence the reaction is by no means complete.
Annealing temperature
The annealing temperature is one of the most critical parameters. Although many individual genes could be amplified at 56°C to 60°C, our experience confirmed that lowering the annealing temperature by 3°C to 4°C was required for the same gene to be coamplified in multiplex reactions. At reduced temperatures, although unspecific amplification probably occurs, it is overcome by the concomitant amplification of an increased number of specific genes in the multiplex reaction and thus remains undetectable. Similarly, when many specific genes are simultaneously amplified, the more efficiently amplified gene will negatively influence the yield of product from the less efficient gene. This is because PCR has a limited supply of enzymes and nucleotides, and all products compete for the same source of supplies.
Number of PCR cycles
Thermocycling parameters are also determined largely by the sequence of the primer sets. Generally, a gradual increase in the yield of all PCR bands with the increase in number of cycles was observed. The most obvious variation in the amount of products was around 25 cycles. Thirty to 35 cycles are usually sufficient for even a multiplex reaction; not too much could be achieved by increasing the cycle number up to 50. Moreover, long extension and annealing times could provide opportunity for nonspecific amplification.
Amount of cDNA template
To rule out variation in the data, it is important to show that the change in cDNA/RNA level does not affect the other in a multiplex reaction. We found that at cDNA template quantities between 0.5 and 1.75 µL per 12.5-µL reaction mixture, there were no significant differences in the amplification efficiency and detection limit of the primers; however, below 0.5 µL, the amount of some of the products decreased.
Primer concentration
In multiplex PCR, adjusting the primer amount for each gene/locus is essential. Initially, equimolar concentrations of 10 µM each were used in the multiplex PCR, but uneven amplification was observed, with some of the products barely detectable even after the reaction was optimized for the cycling conditions. Overcoming this problem required changing the proportions of various primers in the reaction, with an increase in the amount of primers for “weak” genes and a decrease in the amount for the “strong” genes. The optimal concentration of the primers ( Table 1 ) varied considerably among the genes and the sets and was hence established empirically.
Quantitative real-time PCR
Predesigned assays on demand TaqMan® probes and primers were procured from Applied BioSystems (Foster City, CA; see Table 3 ). Total RNA was extracted from undifferentiated hESCs and EBs and reverse transcribed using Superscript II (Invitrogen). Quantitative (q) RT-PCR analysis was performed using the ABI Prism® 7900HT Sequence Detection System (Applied BioSystems). After an initial denaturation for 10 min at 95°C, the reaction was run for 40 cycles of PCR (95°C for 15 s, 60°C for 1 min) per the manufacturer’s protocol. Changes in gene expression (in duplicate) were normalized to 18S rRNA levels.
Confirmation for Differential Expression Levels of Multiplexed Gene Markers in HUES-7 and HUES-9 by Quantitative Real-Time PCR
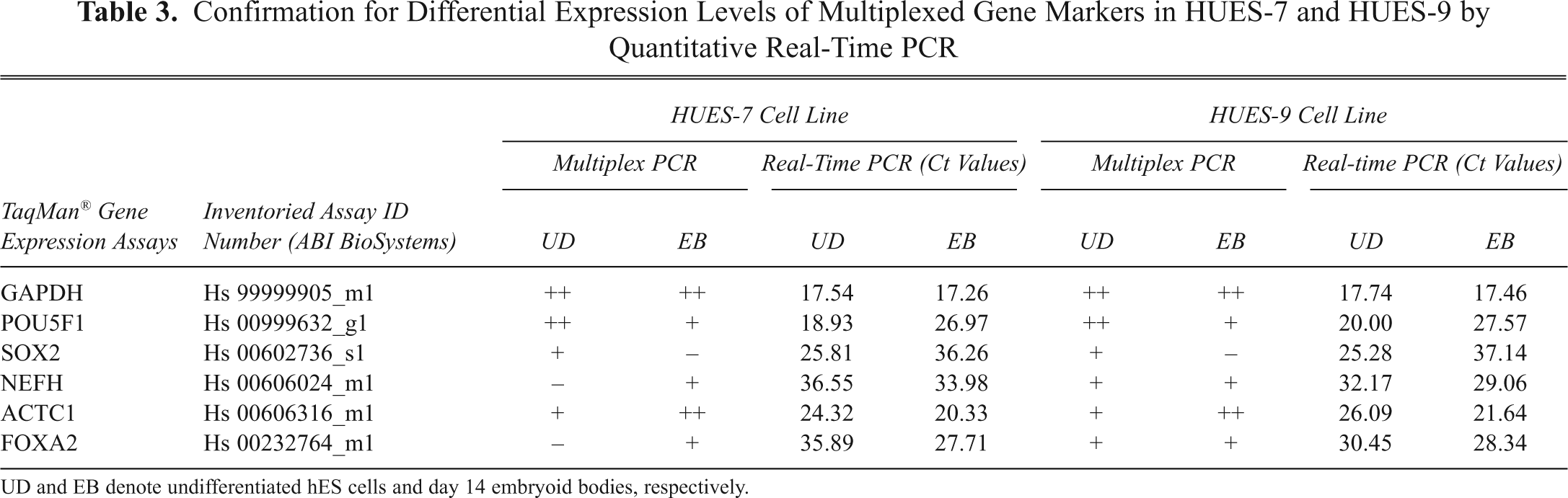
UD and EB denote undifferentiated hES cells and day 14 embryoid bodies, respectively.
Results
Culture and differentiation of hESC and other cell lines
The hESC lines used in this study have been extensively characterized previously, and they express cell surface antigens and relevant transcriptional markers of hESCs as well as exhibit in vitro pluripotency.
3
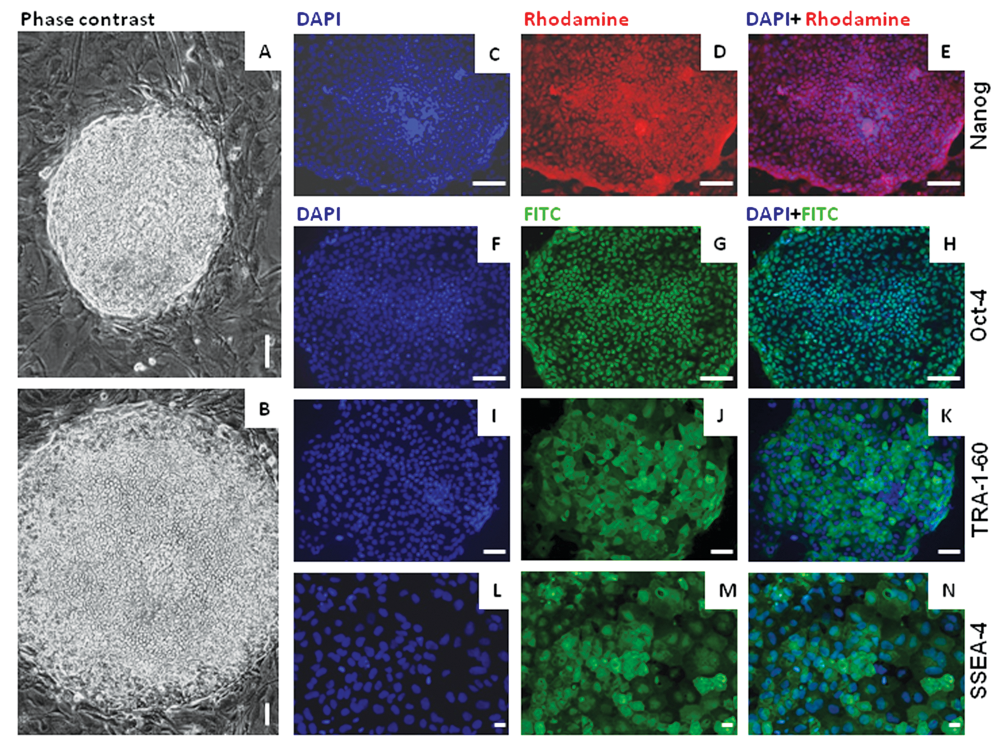
Morphological and immunological characterization of undifferentiated state of human embryonic stem cells. Phase contrast images of compact day 5 undifferentiated HUES-7 colonies under (
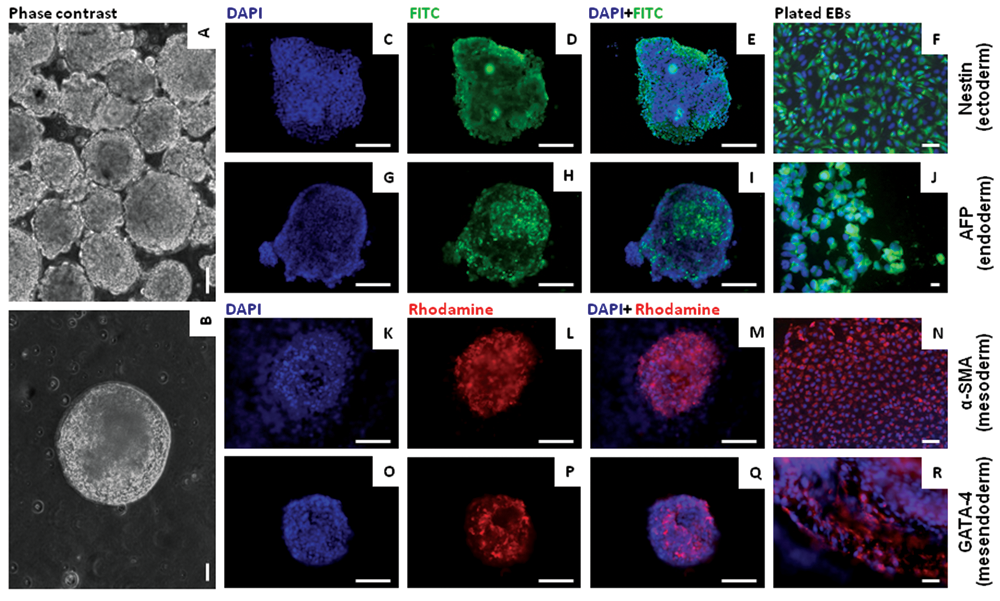
Immunofluorescence analysis of embryoid bodies (EBs). Phase contrast images of (
Expression of stage-specific markers in hESCs
The expression of a set of important cell surface antigens and transcription factors associated with pluripotency and lineage-specific differentiation was examined by immunocytochemistry. Undifferentiated cells and EBs of HUES-7 at passage 38 were stained using antibodies directed against Oct-4, nanog, SSEA-4, TRA-1-61, nestin, α-SMA, AFP, and GATA-4. Representative staining patterns obtained are shown in
Figures 1
and
2
. The results demonstrated that Oct-4, nanog, SSEA-4, and TRA-1-61 markers were expressed by majority of the cells in the hESC colonies (
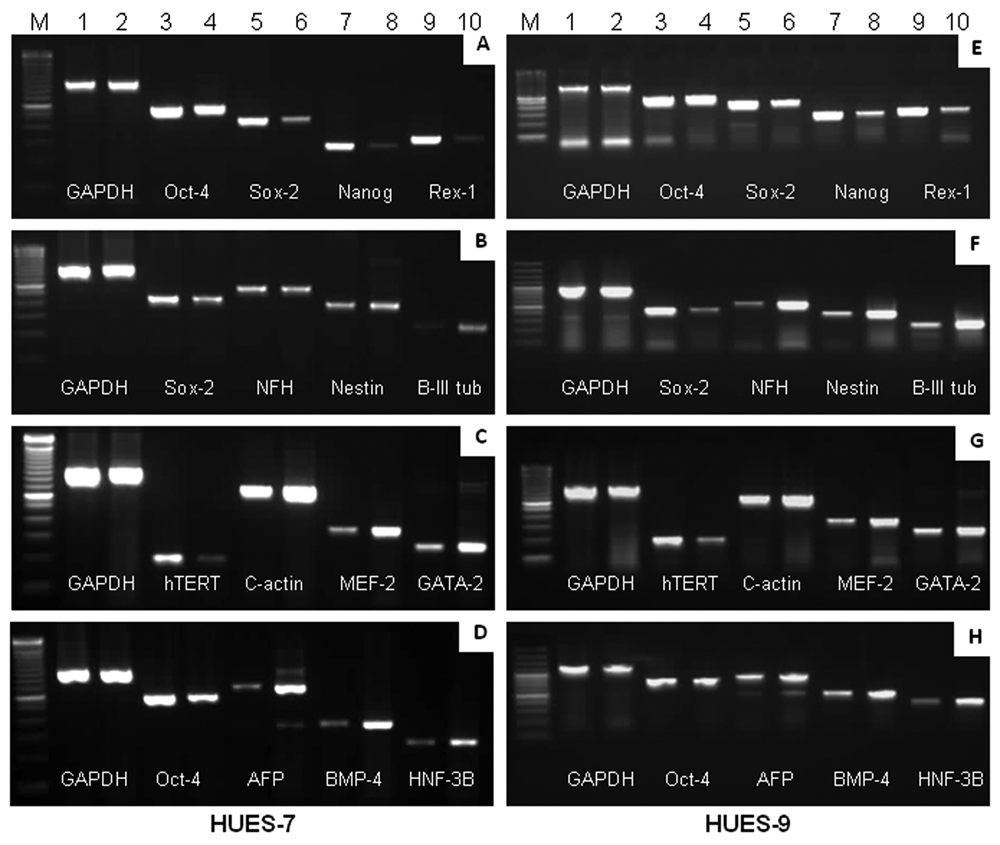
Molecular characterization of undifferentiated human embryonic stem cells (hESCs) and embryoid bodies (EBs) by uniplex PCR. Uniplex RT-PCR analysis of previously reported genes associated with pluripotent state and lineage- or tissue-specific differentiation in both hESC cell lines. (
Development of multiplex PCR and its detection sensitivity
Several permutation and combinations of primers were evaluated for simultaneous amplification of target sequences. After repeated attempts, we have successfully developed the mxPCR technique for 4 separate sets of markers (
Table 1
) using minimal sample volume. Among them, 1 set (set 1;
Fig. 4A
) constitutes pluripotent ESC markers Oct-4, Nanog, Sox-2, Rex1, and the rest of the 3 sets (sets 2-4;
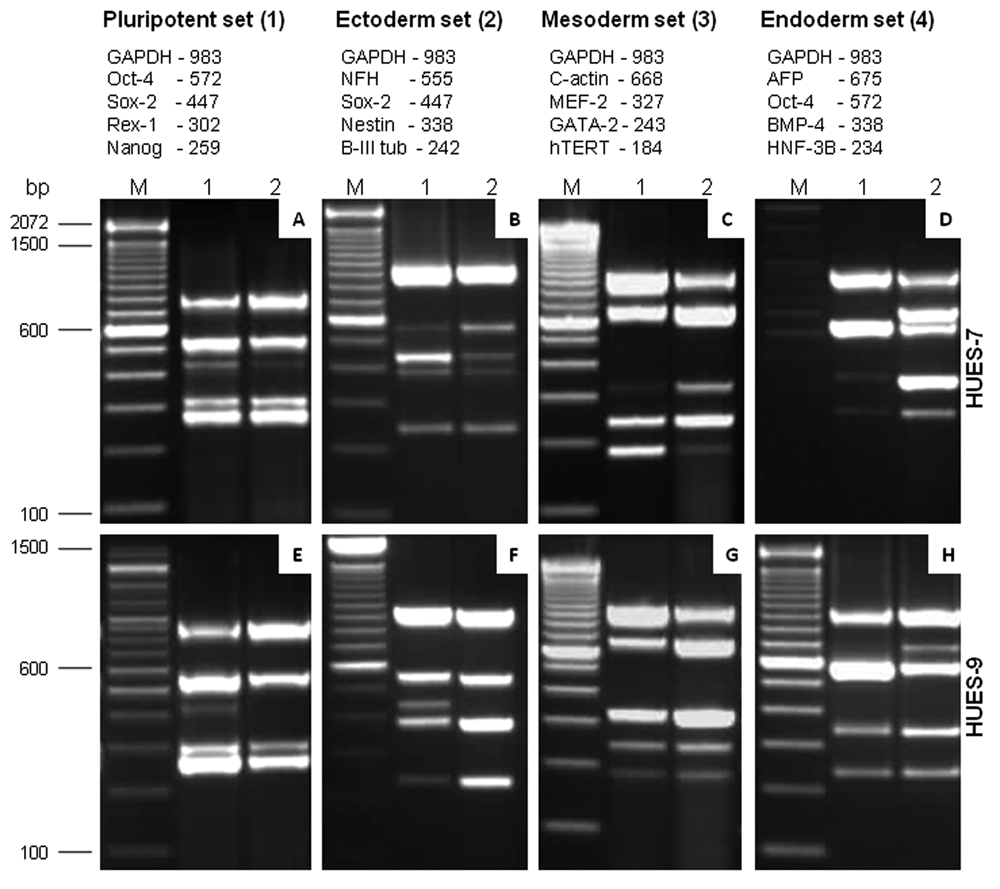
Establishment of mxPCR as a sensitive method for defining the state of human embryonic stem cells (hESCs). Standardization of 4 different sets of multiplex PCRs, wherein each set constitutes a group of prevalidated gene primers representing different stages of hESC differentiation. The name of the gene markers mentioned below follows the order in which they appear in the gel picture from top to bottom. Images
Furthermore, to determine the specificity of this assay and to provide independent verification of the results generated with HUES-7, we performed the same tests using RNA samples of hESCs and EBs from HUES-9 (
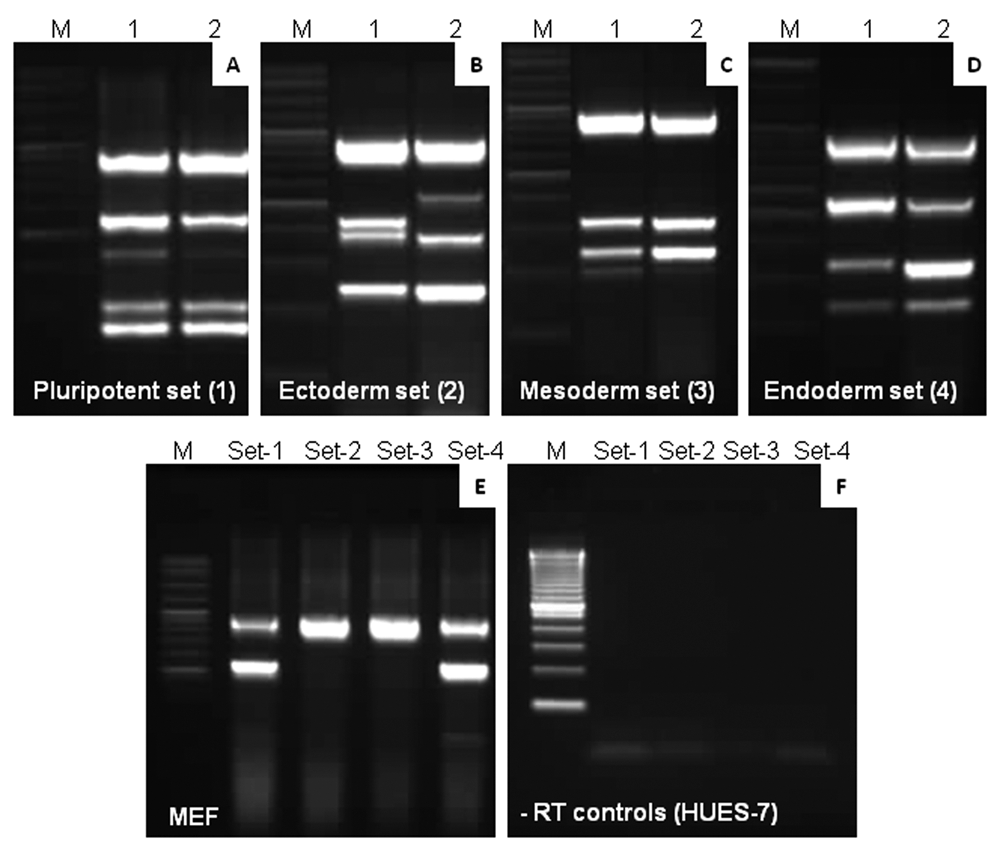
Validation of mxPCR assay using non–human embryonic stem cell (hESC) cell lines. The mxPCR technique we developed was tested for validation in the human teratocarcinoma cell line, NTERA-2, and mouse embryonic fibroblast (MEF) feeder cells. The same gene markers (sets 1-4) and a similar order were followed in this experiment. (
Validation of mxPCR assay
Our results identified an array of 15 genes in 4 different sets that are differentially regulated as cells differentiate to form EBs. Because we standardized the assay with HUES-7 and subsequently reproduced similar results with HUES-9, we validated the same using NTERA-2. Given that HUES-9 and NTERA-2 can easily form neuroectodermal lineage, 3,22 we reasoned that there may be an alteration in the corresponding gene expression levels when compared to HUES-7, which has been shown to have a propensity toward endoderm lineage. 3 We anticipated that our mxPCR assay would be able to detect such small changes in mRNA transcript levels. Likewise, we found that a majority of lineage-specific genes belonging to endoderm in set 4 were downmodulated in day 14 HUES-9 and NTERA-2-derived EBs ( Figs. 4H and 5D ). Moreover, AFP was completely absent in NTERA-2-derived EBs ( Fig. 5D ). On the other hand, in set 2, we identified a distinct upregulation of ectoderm markers such as nestin, β-III tubulin, and NFH ( Figs. 4F and 5A ). Furthermore, in set 3, c-actin could not be detected in NTERA-2 ( Fig. 5C ); nevertheless, there was barely any difference in the levels of pluripotent gene markers ( Figs. 4E and 5A ). As a second criterion, we verified the changes in the levels of gene markers detected in multiplex PCR by real-time PCR analysis and obtained comparable results ( Table 3 ). Here we used a representative set of multiplexed genes for quantitative PCR, which substantiates our claim. This differential display in gene expression indicates that there is a scope of exploring additional differentiation markers to distinguish hESCs at progressive days in differentiation. This finding further indicates that this mxPCR assay is sufficiently sensitive to identify diminutive changes in transcript levels of the proposed set of genes during commitment of hESC toward a particular lineage.
Specific problems encountered during standardization of independent sets
It is important to mention that we developed the PCR conditions separately for each primer set ( Table 1 ). However, we encountered several hurdles during the inclusion of various gene markers conspicuous in ESC such as Oct-4, nestin, β-III tubulin, GATA-2, AFP, and HNF-3β, which were ultimately addressed. In the pluripotent and endoderm sets (sets 1 and 4), GAPDH levels were slightly lower in the undifferentiated hESCs as compared to EBs. We reasoned that because Oct-4 is the most prolific marker for pluripotent ESCs, despite using the same amount of RNA for both samples, Oct-4 emerged stronger to mask GAPDH expression at least to some extent. Moreover, we could not perfect this even by cutting down the primer concentration of Oct-4 considerably. It was also interesting to witness that the specific primer combinations in set 3, such as c-actin, GATA-4, HAND1 or c-actin, MEF2, and HAND1, are not suitable, although the reason remains unclear. In all the differentiation sets (sets 2-4), we successfully combined one pluripotent ESC marker, which led to multiple issues of primer interference and cross-reactivity. However, we took them up one by one and addressed the problems effectively. Furthermore, we noticed that during cDNA synthesis, just before the addition of reverse transcriptase (RT) enzyme, incubating the RNA mix for 2 to 5 min at 42°C improves the quality of cDNA. We speculated this may be due to the time provided for RNA stabilization, which subsequently enhanced the substrate-enzyme reaction kinetics. Finally, we witnessed that the best amplification of our mxPCR products, for the majority of the primer sets, was between 30 and 35 cycles, which was in concurrence with the theory that the probability of nonspecific products is aggravated with an increase in the number of amplification cycles.
Discussion
Unequivocal readouts obtained from gene expression analysis of hESCs at different stages of their development, irrespective of the origin and culture conditions, are important for their successful propagation and expansion in vitro. These data sets also provide significant insights into understanding the differentiation competency of a particular cell line into ecto-, meso-, or endoderm. Preferentially our current strategy of mxPCR is aimed to develop a reliable, robust, simple, and rapid screening method for routine characterization of hESCs. This is precisely of great consequence owing to the recent shift in paradigm from hESC cells to induced pluripotent stem (iPS) cells after realization of the enormous potential of these pluripotent cells in regenerative medicine and drug screening. 23
After several attempts with various combinations of relevant gene primers and an exhaustive regime of troubleshooting, we successfully developed an mxPCR assay that allowed simultaneous and specific detection of a candidate set of markers regularly used for hESC characterization. This report involves a technique in which each primer pair targets a single gene/locus, unlike RAPD 24 or alumorph 25 PCR reactions. There are several advantages of mxPCR over normal uniplex PCR from a practical standpoint (see Fig. 6 ). The most common problem in regular PCR includes false negatives due to reaction failure or false positives due to contamination. False negatives are often revealed in multiplex amplification because each amplicon provides an internal control for the other amplified fragments. The quality of the template may be determined more effectively in multiplex than in single-locus PCR. Evidently, in our study, we used the internal standards of mxPCR to assess the amount of a particular template in a sample. Furthermore, the expense of reagents and preparation time is much less in mxPCR than in systems where several tubes of uniplex PCRs need to be set up. For instance, we have shown cumulative data of 15 different biomarkers ( Table 1 ), including GAPDH as a housekeeping gene, in 4 independent sets categorized on the basis of their function. A multiplex reaction can be ideal for conserving expensive polymerase and templates, which are in poor supply.
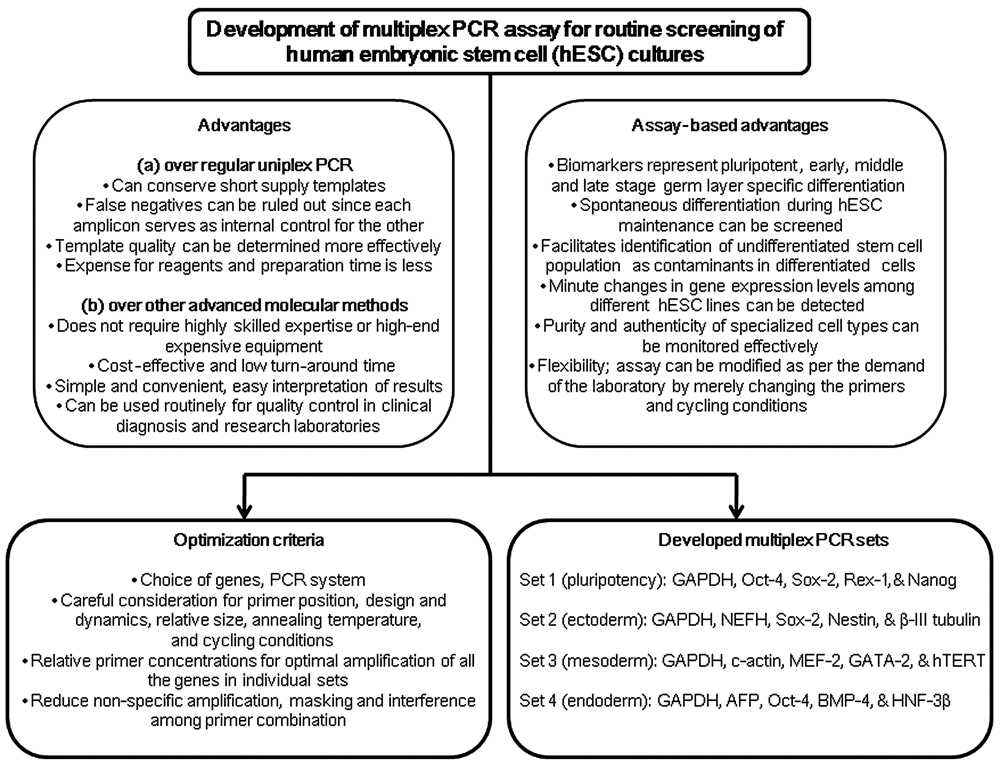
A schematic diagram illustrating step-by-step development of the multiplex PCR assay. This drawing also shows the current challenges of standardization and highlights the superiority of multiplex PCR over uniplex PCR and other advanced techniques of molecular characterization for human embryonic stem cells (hESCs).
Another important facet of this approach is the novel design of the mxPCR. This mxPCR assay was relatively simple as we combined multiple sets of primers for which uniplex PCR reaction conditions were optimized earlier and are routinely in use. 3 However, the mxPCR sets were developed with careful consideration for the specific regions to be amplified, the relative sizes of the fragments, the dynamics of the primers, and the optimization of PCR technique to accommodate multiple fragments. Despite using common primer sets, we encountered several problems while standardizing the PCR conditions for the diverse group of gene markers (cycling conditions, annealing temperature, primer sequence and relative concentration, altering extension times): nonspecific amplification, competition of resources resulting in artifacts, and masking of specific products. Although the unique combinations of markers in sets 1 to 4 offered us more stringent challenges in developing this assay, this was indeed helpful in detecting the amount of undifferentiated cells present in the differentiating population at various time points. Nevertheless, we observed that some primer combinations enhance the possibility of primer complementarity at the 3′ ends, leading to “primer dimers.” These artifacts may deplete the reaction of dNTPs and primers and thereby outcompete the multiplex amplicons for polymerase activity. 26
The significance of subjecting hESCs to routine characterization has also been emphasized by experts in this field. 27 Screening of these cells during in vitro culture at regular intervals is meaningful because they tend to accumulate certain changes in their genomic and epigenomic signature over time. 28 Likewise, even subtle changes in the gene expression pattern of hESCs may alter the pluripotent characteristics of a cell line by modulating its developmental competency. Therefore, it is customary to monitor hESCs during ex vivo expansion at an interval of every 5 passages (maximum 10 passages) more so to avoid cross-contamination when multiple cell lines are used in a particular laboratory. Being simple, efficient, and cost-effective, semiquantitative multiplex RT-PCR analysis may materialize as a convenient option for hESC characterization.
In our earlier report, we demonstrated overexpression of multiple genes in an undifferentiated and differentiated population of hESCs by uniplex PCR, q-PCR, and a microarray technique. 20,21 The present study refines the set of stemness genes to identify those that are rapidly and unambiguously downregulated as ESC undergoes lineage commitment. The selected genes represent a battery of biomarkers to distinguish differentiated cells from pluripotent ESCs. Our results apparently show that many genes that are induced as EB formation starts remain upregulated at days 14 to 21 in differentiation ( Table 3 ). In addition, we witnessed that several markers of early differentiation such as nestin, β-III tubulin, c-actin, GATA-2, AFP, and HNF-3β, which are present in low levels in ESCs, were remarkably upregulated as EBs differentiated. At the same time, levels of pluripotent markers Oct-4, nanog, Sox-2, Rex1, and hTERT were also downregulated. Interestingly, this difference in gene expression between undifferentiated hESCs and EBs when verified employing a real-time q-PCR with a representative set of markers showed reproducible results ( Table 3 ). Hence, we propose that this combination of up- and downregulated gene markers serves as a sensitive indicator of the state of hESC cultures and can effectively be used to monitor the transition of undifferentiated cells into differentiated progenies. However, the observed differences among HUES-7 and HUES-9 may reflect some degree of spontaneous differentiation, an atypical expression of certain markers, or transdifferentiation. For example, we noticed the presence of a small number of differentiated cells of unknown character in our hESC cultures. This is not surprising because manifestation of several pluripotent markers and germ layer–specific markers has been documented earlier in EBs and ESCs, respectively. 18,29,30 However, we found that this faint expression of undifferentiated markers detected in day 14 EBs gradually disappears in 20- or 30-day-old EBs (data not shown).
Among several unique features and benefits of this technique, specifically in stem cell research, the most remarkable is the successful standardization of this assay with half the volume of the PCR reaction mix (12.5 µL) as compared to the common practice of using 25 µL for uniplex PCRs. In this way, we saved precious RNA samples and simultaneously reduced the research expense significantly. The straightforward approach and simplistic methodology of our technique allow the incorporation of additional gene primers depending on the type of cells to be screened. For instance, we were able to replace Sox-2 with TDGF1 in set 1 and c-actin with GATA-4 or GATA-2 with HAND1 in set 2 without changing the PCR conditions (data not shown). Furthermore, we not only detected the presence or absence of important lineage-specific genes (AFP, c-actin) but even could identify miniscule changes in gene expression levels across hESC lines (HUES-7, HUES-9), the human teratocarcinoma line (NTERA-2), and MEF cells. This finding consolidates the sensitivity and robustness of the assay that we standardized. Last, it is noteworthy that we have integrated one pluripotency marker such as Sox-2 in the ectoderm set, hTERT in the mesoderm set, and Oct-4 in the endoderm set, thus facilitating the determination of an undifferentiated stem cell population in differentiated hESC products.
To summarize, we believe that this mxPCR assay may emerge as a promising tool in determining spontaneous differentiation during routine maintenance of hESC and iPS cells. This method permits a clear distinction between undifferentiated and differentiated cells through differential gene regulation. It may also facilitate assessment of a contaminating population of undifferentiated cells in hESC-derived differentiated phenotypes during preclinical or clinical studies. Hence, this assay may be employed as a reliable test for monitoring the purity and authenticity of specialized cells in regenerative medicine and drug screening applications. It is quick, accurate, and sensitive, and unlike other advanced molecular methods, it is affordable, especially in countries with limited economic resources and highly skilled expertise.
Footnotes
Acknowledgements
The authors thank Ms. Greetha Arumugam and Ms. Nor Azah Binti Mori for assistance in cell culture work.
The work was supported by Institute for Medical Research (IMR), Ministry of Health, Malaysian research grant (JPP-IMR-08-019) under Stempeutics Research Malaysia Sdn Bhd. and IMR joint research collaboration.