
Abstract
Lysosomes are membrane-bound subcellular organelles involved in the degradation of macromolecules and pathogens in diverse processes, including endocytosis, phagocytosis, and autophagy. A red fluorescent probe was developed that is selectively sequestered in acidic organelles. U20S cells pretreated with 64 µM chloroquine for as little as 5 h show a dramatic increase in lysosome-like vesicle number and volume. The probe can be employed for highlighting lysosome-like organelles under conditions wherein cells produce vacuoles that contain most of the degradative enzymes of the lysosome but are not as acidic as the parent organelle. Using a conventional fluorescence microplate reader, the half-maximal effective concentration (EC50) of chloroquine was estimated. The high Z′ score obtained using the assay demonstrated excellent signal-to-noise ratios. The fluorescence microplate assay was successfully employed to screen a small-molecule compound library for agents that increase lysosomal volume and number. One potential application of the new assay is in the toxicology portion of preclinical drug safety assessment (ADME-Tox) workflows, using in vitro cell culture models to aid in the drug development process.
Keywords
Introduction
T
The origins of drug-induced lamellar bodies remain unresolved, although they appear to be generated by autophagic or heterophagic processes. 2 Several mechanisms have been proposed to explain drug-induced phospholipidosis, including the formation of drug-phospholipid complexes that are resistant to degradation by lysosomal phospholipases, direct inhibition of phospholipases themselves, and inhibition of intracellular pathways regulating phospholipid metabolism. Operationally, phospholipidosis has frequently been detected in vitro by coincubation of fluorescent analogs of phospholipids with test compounds for extended periods of time. 3-14
We highlight a 96-well cell-based assay that provides a rapid and quantitative high-throughput approach for determining drug- or toxic agent–induced lysosomal perturbation in live cells, offering throughput advantages relative to previously described methods using electron microscopy, fluorescence microscopy, or flow cytometry. The assay is based on incubation of test compounds with cells, followed by brief exposure to a cationic amphiphilic fluorophore tracer that partitions into lamellar inclusion bodies. This tracer was created through careful selection of titratable groups on the probe to enable labeling to be expanded into lamellar inclusion bodies of cells pretreated with weakly basic cell-permeant compounds, such as the antimalarial drug chloroquine. Early secondary screening of candidate drugs for potential lysosome-perturbing activity in the drug discovery phase could predict later risks in drug development arising from drug safety issues. Such a screening approach could aid in selecting a successful candidate compound with low or weak lysosome-perturbing activity for further drug development efforts, as well as provide preliminary benchmarking of dosing limits in preclinical toxicity studies.
Materials and Methods
Materials
The Biomol brand ICCB Known Bioactives Library, Lyso-ID® Red dye, and Hoechst 33342 dye were obtained from Enzo Life Sciences (Plymouth Meeting, PA). Chloroquine, atropine, propranolol, verapamil, imipramine, fluoxetine, chlorpromazine, pimozide, Dulbecco’s modified Eagle’s medium (DMEM) with high-glucose cell culture media, and fetal bovine serum (FBS) were obtained from Sigma-Aldrich Chemical Company (St. Louis, MO). LipidTox reagent, sodium pyruvate, nonessential amino acids, and penicillin/streptomycin solutions were obtained from Invitrogen (Carlsbad, CA). Eagle’s minimum essential medium, McCoy’s 5a modified medium, HeLa cells (ATCC No. CCL-2), MRC-5 (ATCC No. CCL-171), and U20S cells (ATCC No. HTB-96) were obtained from the American Type Culture Collection (Manassas, VA).
Cell culture
U20S human osteosarcoma cells were maintained in McCoy’s 5a modified medium supplemented with 10% FBS and 100 units/mL penicillin/streptomycin in a humidified 37°C, 5% CO2 environment. HeLa human epithelial carcinoma cells were maintained in DMEM supplemented with 10% FBS, 100 units/mL penicillin/streptomycin, and 2 mM glutamine in a humidified 37°C, 5% CO2 environment. MCR-5 human nontransformed fibroblast cells obtained from lung were maintained in Eagle’s minimum essential medium supplemented with 10% FBS and 100 units/mL penicillin/streptomycin in a humidified 37°C, 5% CO2 environment. Although we report extensively on results obtained with human U20S osteosarcoma cell line, the described assay was also validated using the HeLa human epithelial carcinoma and MRC-5 fibroblast cell lines.
Lysosomal perturbation assay
Cells were allowed to proliferate on glass slides or were seeded into 96-well plates at a density of 2 × 104 cells/well and allowed to attach overnight. The cells were plated such that at the end of the experiment, they reached about 90% confluency. Cells were then treated in 8 replicates with each of the compounds tested in the study or with 0.1% (v/v) DMSO, which served as the vehicle control. After 4 to 24 h, cells were incubated for 15 min with a combination of Lyso-ID® Red dye and Hoechst 33342 dye, per manufacturer’s recommendation. Lyso-ID® Red dye is sequestered in lysosomes through careful selection of positively charged titratable groups. The structure and formulation of Lyso-ID® Red dye are proprietary, but it acts through a mechanism that is distinct from commercially available fluorescently labeled phospholipid analogs, such as N-(-7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1, 2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (NBD-PE), N-(-7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1, 2-dihexadecanoyl-sn-glycero-3-phos-phocholine (NBD-PC), and LipidTox reagent. Following incubation, wells were washed 2 to 3 times and analyzed using a fluorescence microscope or fluorescence microplate reader.
Fluorescence microscopy
Cells grown on glass microscope slides were covered with glass coverslips, sealed with nail polish, and observed using an inverted Axiovert 200M microscope (Carl Zeiss, Inc., Oberkochen, Germany). Images were acquired with a 63× objective lens (Zeiss).
Fluorescence microplate-based cytometry
Fluorescence intensity of the cells cultured on the 96-well plates was measured using a FLUOstar OPTIMA Multifunction Microplate Reader (BMG LabTech, Offenburg, Germany) or a Synergy Mx Monochromator-Based Multi-Mode Microplate Reader (BioTek Instruments, Inc., Winooski, VT). Lyso-ID® Red dye and Hoechst 33342 nuclear counterstain can be read with a Texas Red filter set and DAPI filter set, respectively (Hoechst excitation ~300 nm and emission ~480 nm; Lyso-ID® Red dye excitation ~570 nm and emission ~670 nm). Fluorescence intensity was expressed as percentage of the vehicle control value. Values for Lyso-ID® Red dye fluorescence can be normalized to those of Hoechst 33342 dye fluorescence to control for any loss in cell number.
Results
Fluorescence microscopy assay
A fluorescence-based lysosomal perturbation assay was established by first incubating U20S cells cultivated on glass slides for 4 to 24 h with chloroquine (8-64 µM final concentration), a well-known phospholipidosis-inducing agent, and then briefly (15 min) with a combination of Lyso-ID® Red dye and Hoechst 33342 dye. Fluorescence emission intensity and wavelength maximum of Lyso-ID® Red dye are not significantly altered in the pH range of 4.0 to 9.0, allowing interpretation of any increase in fluorescence intensity as an indication of the accumulation of the probe within the cells, rather than as a variation in dye response to lysosomal pH values. Examination of the distribution of the Lyso-ID® Red dye in cells treated with chloroquine revealed a punctate pattern of cytoplasmic staining that increased in a concentration-dependent manner (
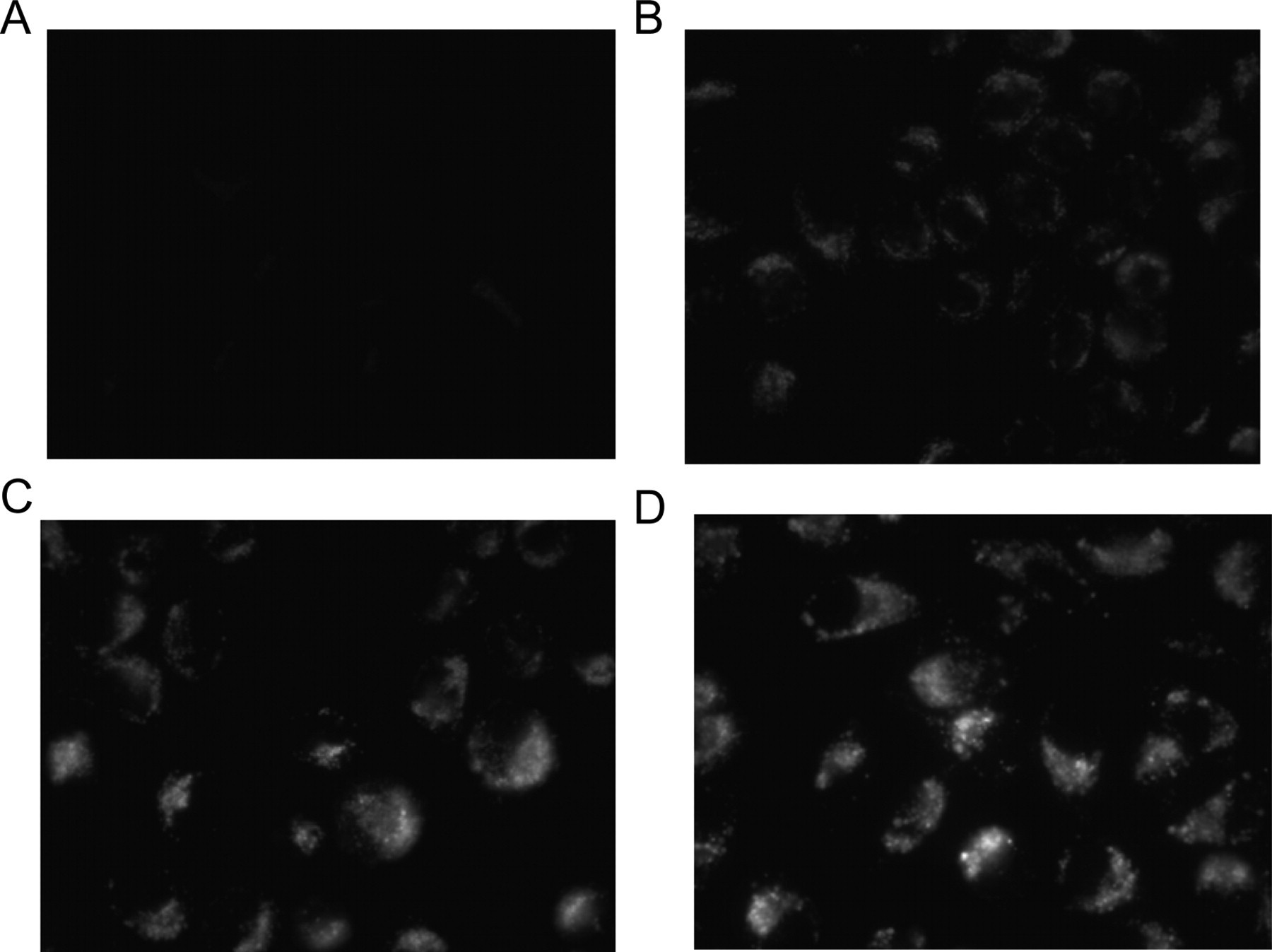
Fluorescent microscopy images of cells treated with increasing concentrations of chloroquine. (
Fluorescence microplate assay
To increase throughput and reduce handling time, the fluorescence microscopy-based lysosomal perturbation assay was scaled up to a 96-well microplate plate assay format and analyzed using a fluorescence microplate reader. Known cationic amphiphilic drugs were used as reference materials in this assay (
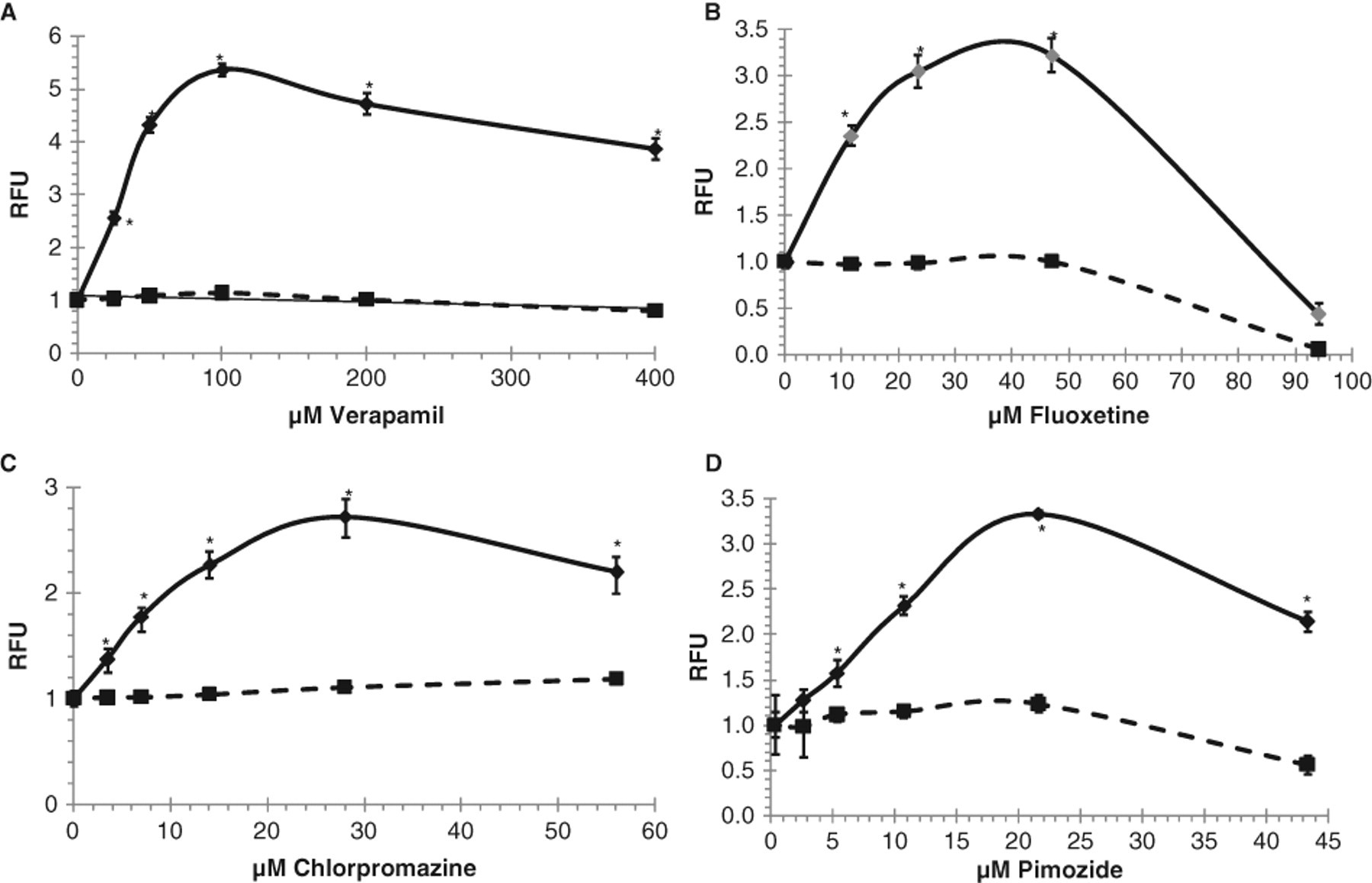
Relative fluorescent intensity of U20S cells treated with known cationic amphiphilic drugs. Cells were treated with (
Relative fluorescent intensity of U20S cells treated with (
When tested in the 96-well assay format using the cited cationic amphiphilic drugs with at least 8 replicates for the test and mock-treated cells, the Lyso-ID® Red dye-based assay demonstrated a Z′ factor value of at least 0.60. This value was determined by comparing signal from the drug concentration giving the highest response, without affecting the Hoechst staining, with the signal from untreated cells. This falls within the range of 0.5 and 1, which is defined as a high-quality assay by the applied statistical parameter. 16 HeLa cells treated with 100 µM verapamil for 19 h in 40 replicates compared with mock-treated cells (40 replicates) also show a Z′ factor value of 0.6. EC50 values for the test drugs in U20S cells ranged from 6 µM for chlorpromazine to 30 µM for verapamil, with chloroquine itself having an EC50 value of 15 µM. In contrast, when using the nontransformed cell line MRC-5, the EC50 for verapamil was 9 µM and for chloroquine was 38 µM (data not shown).
Comparison with a prototypical fluorescent phospholipid analog-based assay
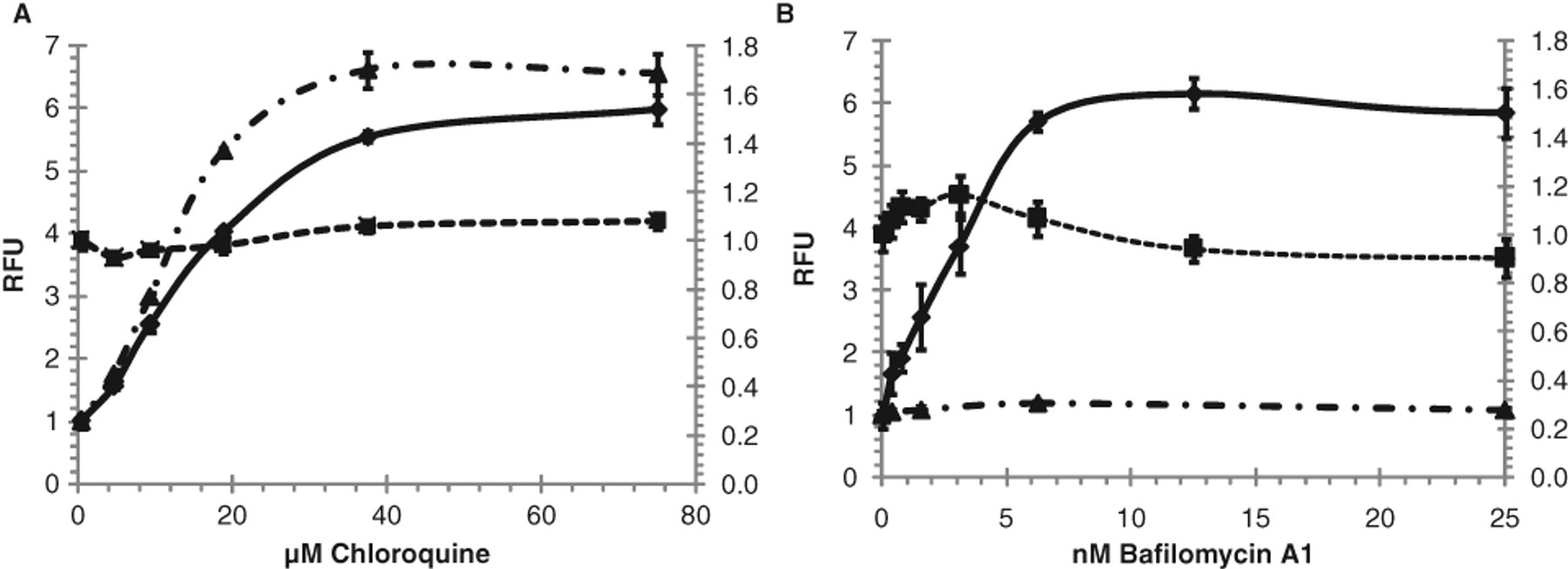
Many assays of phospholipidosis, including the LipidTox assay, are based on coincubating cells with a fluorescent phospholipid analog for an extended period of time, along with the drug or test agent being evaluated, and then monitoring signal generation in fixed and permeabilized cells. The described novel red fluorescent probe is not a fluorescent phospholipid analog but instead is a cationic amphiphilic fluorophore tracer. That is, the tracer is structurally similar to a cationic amphiphilic drug and contains no phospholipid moiety. Cells are incubated with the drug or test agent in isolation and then briefly treated with the tracer dye before evaluation in live cells. This prompted us to speculate that because the 2 assays are based on fundamentally different mechanisms, they might also differ somewhat in terms of the types of test agents that elicited a positive response. Chloroquine, a prototypical phospholipidosis-inducing agent, and bafilomycin A1, an agent well known to induce the accumulation of autophagosomes, were evaluated using both the Lyso-ID® Red and LipidTox dye-based assays (
Compound library screening
As proof of principle, U20S cells were incubated with the 480 compounds in the ICCB known bioactives compound library (Enzo Life Sciences) overnight at concentrations of 0.1 to 53 µM (1000-fold dilution of the stock) and then evaluated using the described lysosomal perturbation assay. In the preliminary screen, test agents were employed at a single concentration in an effort to identify any potential lysosome-modifying agents. Our objective was to benchmark the assay workflow, not to exhaustively classify all of the compounds in the library. It would be advisable to use a range of concentrations for each test agent to reduce the number of false-negative results obtained with the described assay.
The compounds listed in
Positive Hits from a Screen of the ICCB Known Bioactives Library

U20S cells were treated with the indicated compound at the given concentration for 18 h. Compounds that increased fluorescence from Lyso-ID® HSC red detection reagent by greater than 3 standard deviations from the untreated cells are shown. Relative Lyso stain is the intensity of the Lyso-ID HSC red detection reagent fluorescence of treated cells compared with the untreated control. Relative Hoechst stain is the intensity of the Hoechst 33342 fluorescence of treated cells compared with the untreated control, which indicates the number of cells remaining. Compounds listed in bold do not decrease the signal from Hoechst less than 70%.
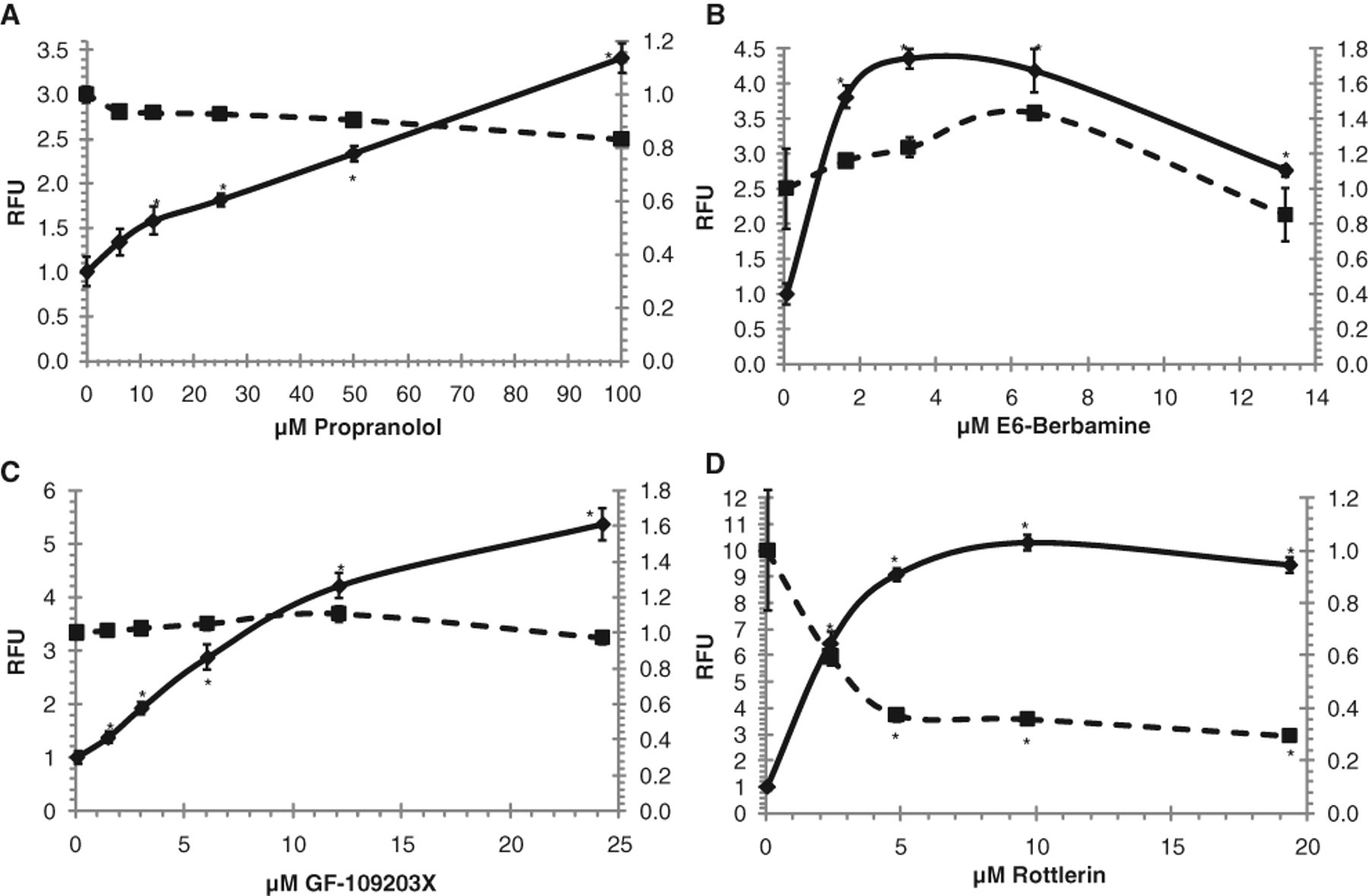
Relative fluorescent intensity of U20S cells treated with (
Discussion
We describe a simple 96-well microplate-based live-cell assay that sensitively detects drug-induced changes in lysosomal number and volume. Numerous in vitro methods for detecting phospholipidosis using different cell types, including primary hepatocytes, peripheral blood monocytes, and various cells lines, in combination with electron microscopy, flow cytometry, fluorescent microscopy, or spectrofluorometry, have previously been described. 1,3-14,17 However, many of these in vitro methods are not ideal for routine compound screening during early stage drug development because of their inherent low throughput. For example, electron microscopy, fluorescence microscopy, and flow cytometry are generally considered low-throughput platforms for routine drug screening purposes, especially when contrasted with fluorescence-based multiwell microplate assays. In addition, some of the above-cited assays depend on difficult to procure cells. For instance, the preparation of primary hepatocytes or peripheral blood monocytes is too laborious for routine drug screening assay programs.
Many of the described cell-based methods are suitable for a plate assay but require a relatively long period of treatment with the test agent before phospholipidosis can be detected (24-72 h). In addition, the vast majority of the assays are based on fluorescently labeled phospholipid analogs, such as NBD-PE, NBD-PC, and LipidTox reagent, which require coincubation of the analog with the drug during the extended incubation period. However, distinct types of phospholipids appear to accumulate differently during phospholipidosis, and no explicit comparison of the performance of the different fluorescent lipid analogs has been performed to date. 18 Unlike the cited phospholipid analog-based probes, Lyso-ID® Red dye is a cationic amphiphilic fluorophore tracer that rapidly partitions into cells in a similar manner as drugs that induce phospholipidosis. Cells are incubated with the test agent alone and then stained for 15 min with the dye. Unlike the fluorophore analog-based approaches, which require fixation and permeabilization to reduce dye background fluorescence, Lyso-ID® Red dye–stained cells may be analyzed directly without these additional processing steps. As a note of caution, fixation and permeabilization are in fact not recommended for the assay, as they lead to rapid dissipation of the dye signal.
We are aware of only one instance wherein a cationic amphiphilic fluorophore has been employed for monitoring drug-induced phospholipidosis. 17 Lysotracker Red DND-99 (Invitrogen) was used to stain lysosomes, and inhibition or displacement of the accumulated dye by test agents was subsequently monitored by fluorescence microscopy. The Lyso-ID® Red dye–based assay is a direct measure of a drug’s ability to induce phospholipidosis in vitro that requires no exogenous coincubation with modified phospholipids. As such, it can be considered a more straightforward and easily interpretable method for assessing phospholipidosis-mediated cell damage than methods based on dye displacement or fluorescent lipid analogs.
Small-molecule activators and inhibitors are commonly used to provide insight into both the mode of regulation and physiological roles of various subcellular targets. However, more extensive screening of these compounds generally reveals that most have overlapping specificities, complicating interpretation of their overall effects on biological systems. Our screen of the ICCB compound library uncovered 5 compounds that substantially increase lysosomal number and volume. Two of these agents, propranolol and GF-109203X, have previously been reported to induce phospholipidosis in cells.
Propranolol is a well-known and extensively investigated cationic amphiphilic drug that induces phospholipidosis. 17,19 High micromolar concentrations of the drug are rapidly taken up and accumulated by a wide range of cultured cells, with a steady state being reached after 40- to 60-min administration and half-maximum uptake within 4 to 10 min. The rate of propranolol dissociation is much slower than the rate of uptake, with 10% of the propranolol remaining associated with the cells after a 90-min incubation period in the absence of the drug. 19
In a recent cell-based high-throughput screen for anthrax toxin inhibitors, GF-109203X was identified as an agent displaying both protein kinase C inhibitory and lysosomotropic activities. 20 Screening of bisindolylmaleimide compounds related to GF-109203X, which lack protein kinase C inhibitory activity but retain tertiary amines and thus the potential to neutralize endosomal pH, was subsequently undertaken by the investigators. This established that the observed toxin inhibitory activity correlated strongly with the presence of the tertiary amines but not with protein kinase C inhibition. 20 In our own screen, Ro 31-8220, a bisindolylmaleimide compound closely related to GF-109203X, but not to our knowledge previously described as a lysosome perturbation agent, was also found to be a positive hit. A structurally unrelated protein kinase C δ inhibitor, rottlerin, increased subcellular accumulation of Lyso-ID® Red dye but displayed significant toxicity, as demonstrated by loss of Hoechst 33342 dye signal in the assay. Reexamination of this compound over a broad range of concentrations revealed it to be generally cytotoxic (data not shown). Intracellular accumulation of Lyso-ID® Red dye in treated cells was always diffuse and cytoplasmic, demonstrating it to be a false-positive hit. Our results, taken together with that of Sanchez et al., 20 suggest that the observed accumulation of Lyso-ID® Red dye in vacuoles correlates strongly with the cationic amphiphilic nature of the bisindolylmaleimide compounds, rather than any protein kinase C inhibitory activity.
Two other positive hits in our screen were SB-431542 and berbamine. SB-431542 is reported to be a transforming growth factor-β-receptor antagonist that serves as a potent, competitive, adenosine triphosphate (ATP) binding site kinase inhibitor for the downstream activin receptor-like kinases, ALK4, ALK5, and ALK7. 21 Physiologically, SB-431542 has previously been shown to inhibit cell proliferation and block cell motility. Berbamine is reported to selectively induce caspase-3-dependent apoptosis of leukemia NB4 cells via a survivin-mediated pathway. 22 Our subsequent evaluation of the structurally related alkaloid, isotetrandrine (aka berbamine methyl ester), demonstrated that it too dramatically increased lysosome number and volume. We are not aware of any previous reports that these agents have a propensity to induce vacuolation in cells, and indeed their chemical structures are not consistent with them belonging to the cationic amphiphilic class of compounds.
In addition to authentic compounds that cause phospholipidosis, agents that cause the accumulation of autophagosomes by blocking the downstream lysosomal pathway and/or intracellular trafficking of autophagosomes may also lead to increases in the accumulation of intracellular Lyso-ID® Red dye signal in the described assay. For example, bafilomycin A1, a macrolide antibiotic isolated from Streptomyces griseus, is a particularly potent agent in our assay. Initial screening of this compound using the ICCB library revealed it to be cytotoxic, but rescreening at much lower concentrations demonstrated its ability to increase vacuole number and volume. Bafilomycin A1 is a rather specific vacuolar type H+-ATPase inhibitor that is known to increase the numbers of intracellular autophagosomes, purportedly by blocking the ability of the lysosome to degrade them. 21,23 The bafilomycin A1 results suggest that the described assay should be considered a measure of generalized dysfunction of the lysosomal-dependent catabolic degradation pathway, which leads to the accumulation of autophagosomes, and thus relevant to autophagy in general, as well as phospholipidosis in particular.
Footnotes
The authors are employed by the company that manufactures and sells the kit described in this publication.