
Abstract
The authors describe a technique for mapping the epitopes of protein antigens recognized by mono- or polyclonal antibodies. This method is based on a recombinant polypeptide library, expressed in a bacterial expression system, arrayed at high density, and tested on a membrane with automated procedures. The authors analyzed the epitope of a commercially available monoclonal antibody to vitamin D receptor (VDR). About 2300 overlapping VDR peptides were screened on a test array, and a contiguous stretch of 37 amino acids was identified as the epitope. Its authenticity was confirmed by Western blotting and an immunofluorescence competition assay on human skin tissue samples. The authors define the proposed method as a cell-based protein or peptide array that is adaptable to many applications, including epitope mapping of antibodies and autoantibodies, autoantigen detection from patient sera, whole-proteome approaches such as protein-peptide interactions, or selection of monoclonal antibodies from polyclonal sera. The advantages of this method are (a) its ease of protein array production based on well-established bacterial protein/peptide expression procedures; (b) the large number of printable colonies (as many as ~25,000) that can be arrayed per membrane; (c) there is no need for protein purification of recombinantly expressed proteins; (d) DNA, rather than protein, is the starting material to generate the arrays; and (e) its high-throughput and automatable format.
Keywords
Introduction
T
Protein arrays can be classified as either cell-free or cell-based arrays displaying real or synthetic polypeptides. Cell-based arrays, especially ones for human cells, are often limited to a few hundred probes. 8,9 Cell-free protein chips produced by using an in vitro translation system 10 or an in vitro transcription and translation system 11,12 are now available, thus overcoming the labor-intensive protein preparation procedure in protein-array construction. 13 A second well-known cell-free method for protein chips is the SPOT-synthesis technique, 14 where mainly peptides are spotted onto a membrane. This method is limited by the need to reduce the spot size to below 1 mm and becomes costly when large numbers of the same array are required. 15 Therefore, the process for manufacturing synthetic peptide arrays requires additional effort and expense. For simplicity, we use the term protein array to refer to a large number of different polypeptides displayed on a surface (membrane or chip) irrespective of whether the polypeptides are short, long, or protein length and irrespective of the means of array fabrication, whether spotted as protein or expressed in cells and lysed.
Many methods for mapping epitopes have been invented. Which method to use depends mainly on the available resources. The most precise method is to determine the atomic structure of an antigen-antibody complex by x-ray crystallography or nuclear magnetic resonance. 16-19 However, neither technology is routinely used for this purpose because of their high costs, time demands, and limited utility for investigating large protein complexes. Two more general technologies are more commonly used. The first involves enzymatic cleavage of the antigen and mass spectrometric analysis of the cleavage products. 20 The antigen can be investigated alone and the digested fragments captured by the antibody (epitope extraction), or the antigen-antibody complex can be investigated in toto, in which case antibody binding prevents the epitope from being enzymatically cleaved (epitope excision). 21,22 A second common tool uses synthetic peptides. In this approach, the amino acid sequence of the antigen must be known. An example is the widely used “Geysen pepscan” method introduced by Mario Geysen. 23 In this technique, short, overlapping synthetic peptides are tested for their ability to bind the antibody. Depending on the peptide sizes and the required number of peptides (a function of protein size), this can be a very laborious and costly effort.
Here we describe a general method for mapping the epitopes recognized by specific antibodies, by generating and probing a high-density protein array. With this technique, screening of arrays containing recombinant peptides is possible. As a demonstration of this approach, we describe a high-throughput epitope mapping screen of a recombinant peptide library. A total of 2304 overlapping peptides of the vitamin D receptor (VDR) expressed on a protein array were probed with an anti-VDR monoclonal antibody, permitting identification of a 37–amino acid continuous epitope of VDR. Other applications of the method include the directed production of monoclonal antibodies out of polyclonal ones. For example, the mapped polyclonal epitopes could be used for immunization of production animals and manufacturing of monoclonal antibodies. Furthermore, the mapping of antigens in autoimmune diseases is of great interest in clinical research and diagnostics and is facilitated by the use of protein chips or protein arrays. 24 Epitope mapping is an important step in characterizing the autoantibodies that bind to the autoantigen, a potential therapeutic target. 25,26
Materials and Methods
Library construction
The human VDR gene encodes a member of the nuclear receptor family of transcription factors that plays a key role in binding nutritionally derived ligands that exert effects that contribute to bone mineral homeostasis. 27 The nucleic acid sequence of human full-length VDR cloned in pBD-Gate2 vector 28 was amplified with gene-specific primers (forward: 5′-ATGGAGGCAATGGCGGCCAGCA-3′; reverse: 5′- AGGAGATCTCATTGCCAAACA-3′). The resulting 1.2-kb PCR product was purified with a Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI) according to the kit’s instructions and then fragmented by sonication. Separation of the sonicated DNA on an agarose gel showed fragments down to 100 bp. The fragments between 100 and 300 bp were purified again and blunt-ended by using the End-It™ DNA End-Repair Kit (Epicentre, Madison, WI). To obtain flexibility in manipulation, we cloned the fragments into an in-house-created Entry vector designed for site-specific recombinational cloning. 29,30 To achieve this, the vector pDONR™/Zeo (Invitrogen, Carlsbad, CA) was equipped with a restriction site for blunt-end cloning. After ligation into pDONR™/Zeo and attL × attR-directed recombination 30 into the bacterial expression vector pDEST™15 (Invitrogen), 1 × 106 individual clones were obtained.
Creation of the protein array, colony spotting, and recombinant protein expression
The library in pDEST™15 was used to transform Escherichia coli BL21 Star™ (DE3) competent cells (Invitrogen), and the cells were plated onto LB medium plates containing ampicillin (100 µg/mL). A total of 2304 colonies were picked by a QPix2XT robot (Genetix, New Milton, Hampshire, UK) into 384-microwell plates containing liquid LB medium supplemented with ampicillin (100 µg/ml). After cultivation, the cultures were spotted robotically onto a PROTRAN™ nitrocellulose transfer membrane (Whatman GmbH, Dassel, Germany) with a pore size of 0.45 µm. Cells were grown overnight at 37 °C on this membrane overlaid on solid LB medium. Protein expression was induced by transferring the membrane onto solid LB medium supplemented with ampicillin and isopropyl β-
Epitope screening and detection
The cells on the membrane were lysed by transferring the membrane into a phosphate-buffered saline (PBS) solution containing 0.02% Tween-20 and 3% blotting-grade milk powder (Carl-Roth GmbH, Karlsruhe, Germany), and the released recombinant proteins became bound to the nitrocellulose. For detection, a refined dot-blot procedure was used, which was scaled up to accommodate a 22.2 × 22.2-cm2 large membrane and fluorescently labeled secondary antibodies and fluorescence scanning on an Amersham Typhoon™ Scanner (Amersham, Pittsburgh, PA). The primary antibody was a commercially available mouse monoclonal IgG2A antihuman VDR antibody (sc-13133; Santa Cruz Biotechnology, Santa Cruz, CA). For detection, an Alexa Fluor® 488–conjugated goat antimouse IgG antibody (Invitrogen) was used. Array spots representing positive antibody binding were detected with the Amersham Typhoon™ Scanner. Excitation occurred at 532 nm, and emission was measured with a 526-nm filter. Only spots that delivered a fluorescence signal at least 100% above background were considered as hits and subjected to DNA sequencing. We defined the background by calculating the mean of 10 randomly chosen nonfluorescent spots.
DNA sequencing and analysis
Bacterial cultures of cells representing positives spots on the protein array were subjected to plasmid isolation. A vector-specific primer (5′-GGCAAGCCACGTTTGGTG-3′) was used for standard dideoxy sequencing 31,32 with an ABI 3130 Genetic Analyzer Capillary Array (Applied Biosystems, Foster City, CA). The resulting sequences were inspected and aligned. The minimal overlapping sequence was taken for further validation.
Western blotting and immune fluorescence
The DNA sequence encoding the 37–amino acid epitope was made Gateway® compatible by using first specific and then secondary adapter primers 33 and then cloned into pDONR™/Zeo (Invitrogen). After attL × attR recombination, the minimal sequence was shuttled into pDEST™15 (Invitrogen). First, the pDEST™15 epitope construct was used to transform E. coli BL21 Star™ (DE3) cells (Invitrogen), and recombinant protein expression was induced in the exponential growth phase for 2 h with 0.5 mM IPTG. The recombinant proteins were purified by using a MagneGST™ Protein Purification System (Promega) according to the kit’s protocol. For Western blot procedures, the purified protein and a crude lysate were used for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The same anti-VDR antibody was used. As a secondary detection antibody, alkaline phosphatase–linked goat IgA antimouse antibody (Sigma-Aldrich, St. Louis, MO) was used. An in-house-created pDEST™15 vector harboring a stop codon was used to prepare a negative control lysate, and lysate from cells expressing full-length VDR in pDEST™15 was used as positive control. As a validation step, the purified epitope was preincubated with the anti-VDR monoclonal antibody to saturate the epitope binding sites, and as a control, an equal amount of nonblocked anti-VDR monoclonal antibody was used. Six-micron-thick cryosections of human skin tissues were washed in PBS (pH 7.4), fixed with 4% formaldehyde in PBS for 10 min, permeabilized with 0.25% Triton X-100 (AppliChem, Darmstadt, Germany) in PBS for 5 min, and finally washed 3 times in PBS for 5 min. The skin sections were probed with the blocked and nonblocked anti-VDR monoclonal antibody separately overnight at 4 °C. After another washing step in PBS for 5 min, Alexa Fluor® 594–labeled goat antimouse IgG antibody (Invitrogen) was used as the secondary detection antibody in a 1-h incubation at 4 °C. After a 5-min wash in PBS, the sections were finally treated for 10 min with 4′,6′-diamidino-2-phenylindole (DAPI; dilution 1:2000 in PBS; Sigma-Aldrich) and examined with a Zeiss Axioskop MC100 (Carl-Zeiss Micro Imaging, Göttingen, Germany).
Enzyme-linked immunosorbent assay
To detect specific binding of the commercial anti-VDR antibody to the pDEST™15 epitope construct, we performed a standardized enzyme-linked immunosorbent assay (ELISA). For this, the pDEST™15 epitope construct as well as the positive (pDEST™15 VDR full-length) and negative (pDEST™15 Stop) controls were bacterially expressed and purified using the MagneGST™ Protein Purification System (Promega) according to the kit’s protocol. Each well of the 96-well microplate (Nunc Polysorb™, Rochester, NY) was coated with 1 µg of epitope or control proteins in 100 µL of sterile-filtered ELISA/ELISPOT coating buffer (eBioscience, San Diego, CA). The plates were incubated overnight at 4 °C followed by washing thrice with distilled H2O. Nonspecific binding sites were blocked by adding to each well 200 µL of 3% bovine serum albumin (BSA) in PBS containing 0.02% Tween-20 (PBST) and incubating at room temperature for 1 h. After the blocking step, the wells were incubated with 100 µL of primary antibody at a concentration of 0.4 ng/µL in PBST containing 3% BSA at room temperature for 2 h. The first antibodies used here were the epitope-mapped mouse IgG2A monoclonal anti-VDR antibody, a mouse IgG2A monoclonal anti-RACK1 (receptor for activated C kinase 1) antibody (sc-17754; Santa Cruz Biotechnology), and normal mouse–IgG (sc-2025; Santa Cruz Biotechnology). Each antibody was tested in triplicate against the epitope and the controls. As a further control, just 3% BSA in PBST was applied to wells to detect possible binding of the secondary antibody. After washing 3 times with PBST, the plates were incubated with 100 µL per well of horseradish peroxidase (HRP)–conjugated goat polyclonal to mouse IgG (Abcam, Cambridge, MA) at a concentration of 2 ng/µL diluted with 3% BSA in PBST for 1 h at room temperature in the dark, followed by 5 final washes each with 200 µL PBST. As a substrate, 200 µL per well of dissolved SIGMAFAST™ OPD (o-phenylenediamine dihydrochloride) tablets (Sigma-Aldrich) was used. To detect background activation, empty wells were incubated with the substrate, too. The plate was developed for 60 min in the dark, and the absorbance of the contents of each well was read at 450 nm in a PARADIGM™ detection platform (Beckman Coulter, Brea, CA). The absorbance (optical density, OD) of each well was determined by subtracting the mean of the 5 background test wells. Each epitope/control combination with the antibodies was done in triplicate. The mean OD was used to express the measurement.
Results
An outline of the basic principle is given in Figure 1 . In summary, we have created a facile, automated method for epitope mapping.
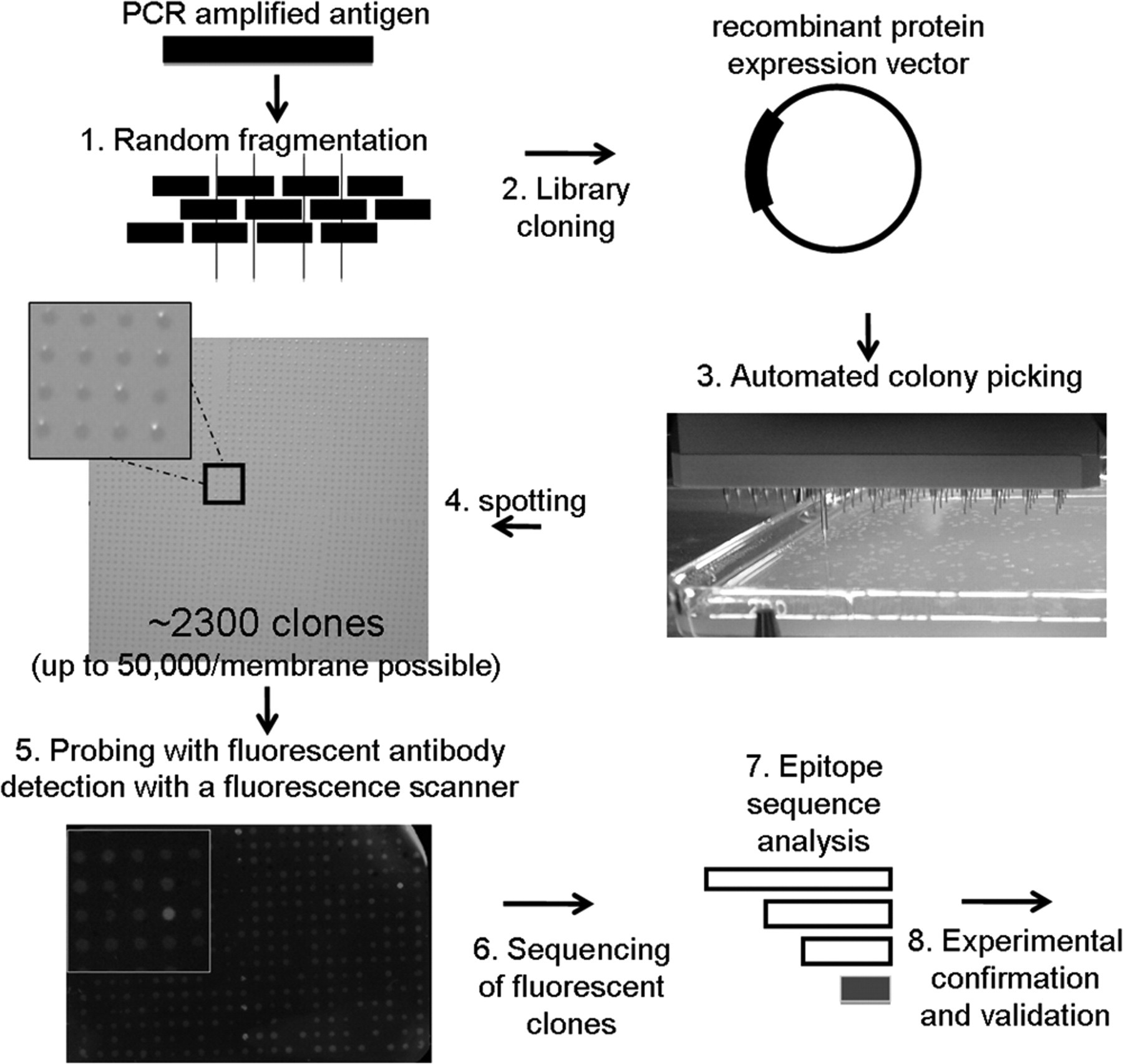
Schematic of the epitope mapping procedure. The antigen encoding gene is PCR-amplified and fragmented by sonication (1). The ends are repaired and cloned by ligation followed by recombination into an Escherichia coli glutathione-S-transferase (GST) fusion expression vector (2). An E. coli expression strain is transformed with the library, and colonies are picked by a QPix2XT (Genetix) robot (3). After cultivation, the cultures are spotted by a robot onto a 22.2 × 22.2-cm nitrocellulose membrane (4). A standard dot-blot protocol is used for fluorescence detection under a Typhoon™ scanner (5). Cells harboring a positive (here a bright spot) are used for plasmid preparation and sequencing (6). The derived sequences are aligned to the antigen for identification of the epitope (7). The minimal overlapping sequence is separately cloned and subjected to epitope validation (8).
Production of a high-diversity human VDR peptide library
For library construction, the full-length VDR open reading frame was PCR-amplified, fragmented by sonication, and cloned into an expression vector for recombinant protein production in E. coli as described in Materials and Methods. Approximately 1 × 106 independent clones were produced, sufficient for a library of a single fragmented gene (see below). The complexity of the created library was estimated by sequencing 30 randomly chosen clones. Approximately 30% of the inserts were 17 to 30 amino acids in length, ~50% were 30 amino acids, and ~20% were 30 to 50 amino acids, all suitable sizes for epitope mapping. The sequencing analysis also showed a cloning efficiency of ~90%. Based on the theoretical assumption that 1 in 6 clones will harbor an insert in the correct frame and orientation (3 reading frames possible in 2 orientations) and a cloning efficiency of 90%, then at least 150,000 correctly inserted in-frame clones should be present in the VDR fragment library. This number of open reading frames should completely cover the human VDR gene numerous times with overlapping inserts.
Identification of a 37–amino acid stretch as the VDR epitope recognized by a monoclonal antibody that is likely linear
For proof of concept, we used our peptide array method to map the known epitope of a commercial antibody against human VDR. The supplier of the antibody states a rough estimate of the epitope region, which is therefore an ideal test system. To produce the protein array, we picked and transferred single colonies into liquid medium-filled 384-well plates with a picking robot and cultivated them overnight at 37 °C. The cultures were automatically spotted onto a 22.2 × 22.2-cm nitrocellulose membrane that is specific for protein and has high affinity for protein binding. A total of 2304 spots were arrayed on the membrane, giving a density of about 4.7 spots per cm2. The cells were allowed to grow on the membrane overnight until they had reached a moderate size, at which point they were directly lysed on the membrane. The lysis method, which uses a weak denaturizing detergent (Tween-20), lysed enough cells to release sufficient recombinant material for detection. We do not believe that the peptides are completely denatured because fluorescence proteins retain their ability to fluoresce after this procedure (data not shown). To screen for antibody epitopes, we subjected the spotted recombinant peptides to antibody detection by a refined dot-blot and fluorescent detection procedure. For evaluation, a secondary fluorophor-labeled antibody was used, and positive spots on the membrane were detected by a fluorescence scanner.
Positive spots were chosen for plasmid preparation and DNA sequencing analysis. According to the sequence analysis, all plasmids prepared from positive spots were found to harbor inserts with in-frame VDR coding sequences correctly fused with the glutathione-S-transferase (GST) tag of the expression vector. Furthermore, comparison of the sequences showed that all the clones encode overlapping parts of 1 distinct VDR region. The full-length VDR protein consists of 427 amino acids. The shortest epitope we are able to define is 37 amino acids, corresponding to residues 391 to 427 of the VDR protein. The manufacturer of the VDR monoclonal antibody cites amino acids 344 to 424 of VDR, an 80–amino acid stretch, as containing the epitope of this antibody. The epitope we determined is found in the same region, which confirms the efficacy of our method. In addition, our method provided more specific information about the epitope region, refining its location from 80 amino acids down to 37 ( Fig. 2 ). This improved characterization of the epitope has practical advantages for VDR antibody production because the shorter peptide is easier and less costly to synthesize.

Sequence alignment of positive candidates. Sequences of isolated positive colonies were translated and aligned to the protein sequence of the full-length human vitamin D receptor (VDR). (
Furthermore, to strengthen our hypothesis that this epitope is linear, we performed 3D modeling of the 37–amino acid segment onto an x-ray crystal structure of the VDR ligand binding domain. A 16–amino acid long C-terminal alpha-helix connected by a 3-residue turn and followed by 14 amino acids of a second alpha-helix comprise the epitope. The last 4 C-terminal residues of the 37–amino acid segment were not included in the crystal structure (
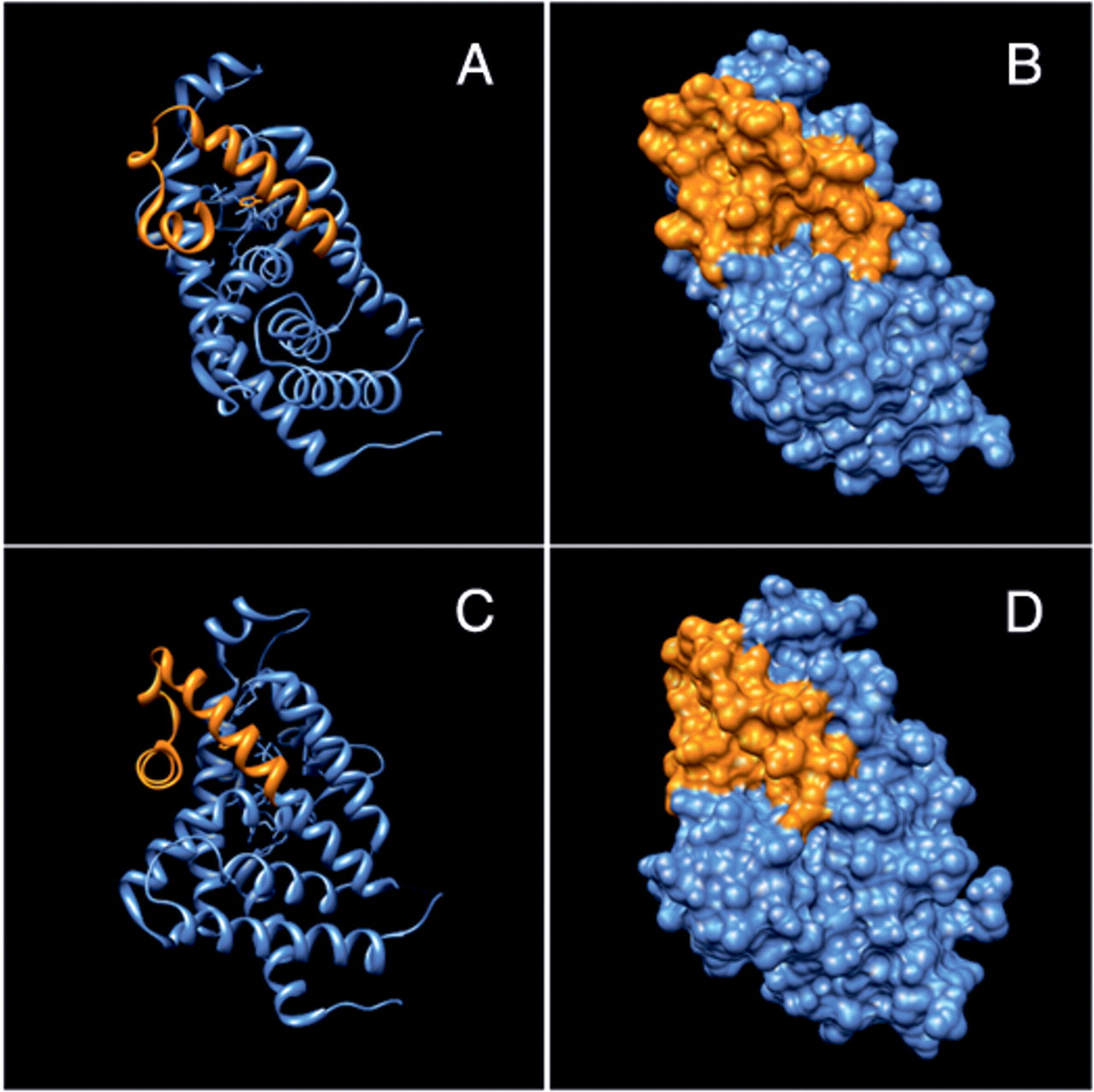
Three-dimensional modeling of the epitope. (
Epitope validation by Western blotting and immunofluorescence competition analysis in human skin samples
To confirm the authenticity of the epitope, we synthesized the corresponding nucleotide sequence as an oligonucleotide and cloned it into the same expression vector as was used for recombinant peptide production. This synthetic epitope was analyzed along with full-length VDR as a positive control as well as an additional negative control by Western blotting with the VDR monoclonal antibody. Reactivity of the expressed short peptide epitope to the monoclonal antibody is shown in Figure 4A .
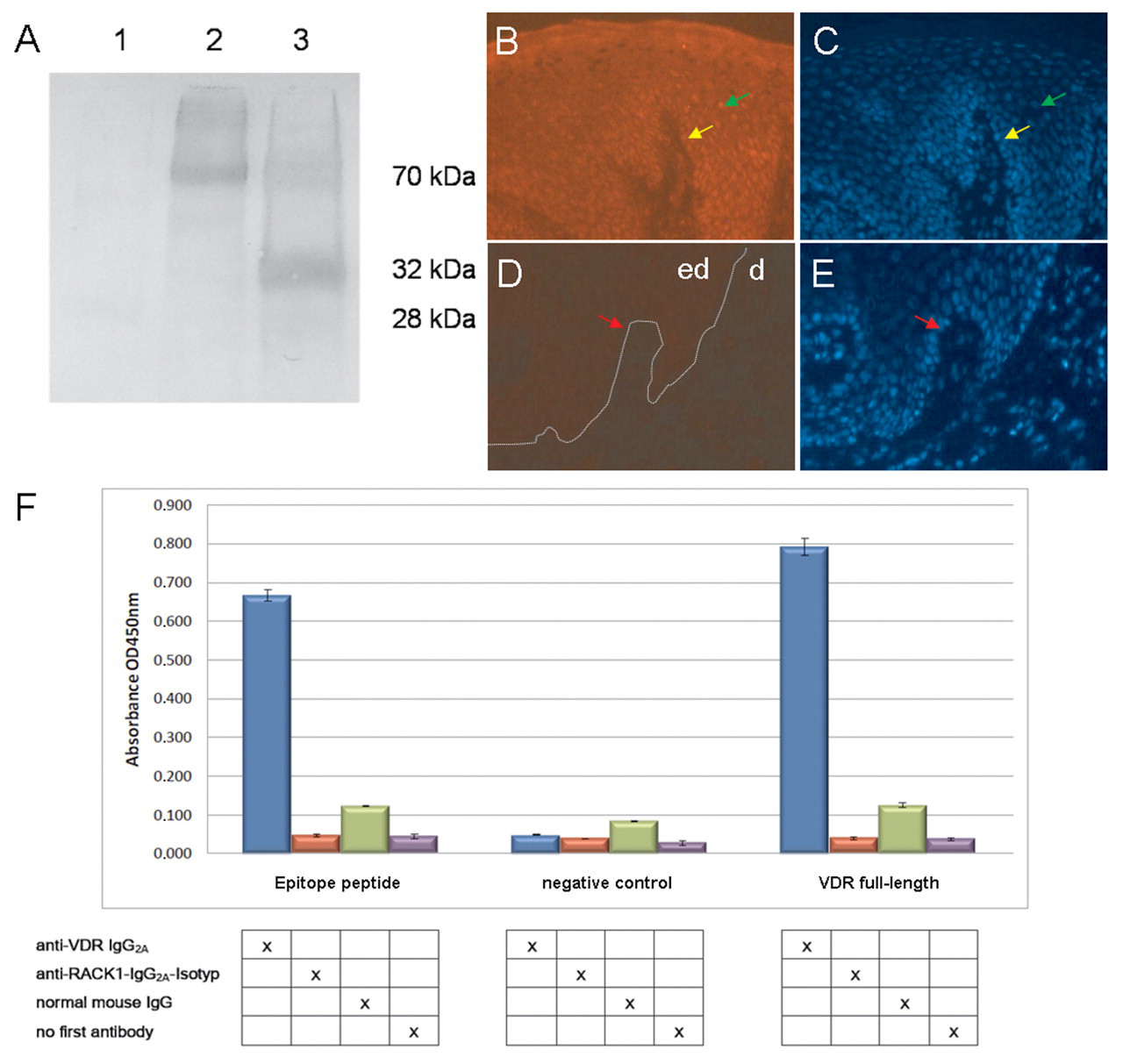
Epitope validation. (
For in vivo validation of the 37–amino acid epitope, we performed an immunofluorescence competition assay. The affinity-purified peptide was able to specifically block the binding of the VDR monoclonal antibody to endogenously expressed VDR in human skin tissues. Preincubation of VDR antibody with the purified peptide reduced the intensity of the signal to VDR in human skin in comparison with an equal amount of pure anti-VDR antibody ( Fig. 4B - E ).
Epitope validation by specific binding of the VDR antibody compared to isotype-matched control antibodies
Another step in epitope validation is to show that the binding of the commercial monoclonal anti-VDR antibody to the epitope is specific and that an isotype-matched control antibody does not bind to the epitope. By ELISA, the VDR IgG antibody, a mouse IgG control antibody, and a mouse anti-RACK1 antibody as a negative control were each tested for binding to (1) the recombinant purified 37–amino acid epitope, (2) a recombinant purified full-length VDR protein, and (3) a negative control (GST tag only). Only the combinations of VDR antibody with the 37–amino acid epitope and VDR antibody with the full-length VDR showed a significant increase in absorbance ( Fig. 4F ). In addition, the controls where no first antibody was used confirmed that the secondary antibody did not bind to the epitopes, producing only background absorbance with the substrate. These results demonstrate specific binding of the VDR antibody to the mapped VDR epitope.
Discussion
The new method described here allows for rapid epitope mapping of mono- or polyclonal antibodies by using a bacterially expressed short-peptide library consisting of overlapping fragments of the antigen and an automated protein array assay. In our method, we combined a cell-based array with a fragmented recombinant peptide library (fragment size range ~17-50 amino acid residues) for epitope mapping, but it can also be used for screening protein interactions with a recombinant polypeptide library of larger inserts (e.g., protein domains or motifs expressed in E. coli cell arrays and screened with antibodies, protein interactors, etc.). We demonstrated the method’s efficacy by mapping the epitope of a commercially available monoclonal antibody to a stretch of 37 amino acids. Moreover, it is likely that the epitope could be further refined to 34 amino acids because the manufacturer’s declared region (amino acids 344-424 of VDR) did not include the last 3 amino acids of the epitope (amino acids 391-427) found in this study. In the case of polyclonal antibodies, the proposed method would make it is feasible to find more than 1 epitope in an antigen. 34 Although we focused on mapping a continuous epitope, mapping of discontinuous epitopes might be achieved by generating a library containing longer fragments, albeit at lower resolution. Moreover, failure to find an epitope in a short-peptide library might be indicative of a discontinuous epitope.
The power of a protein array depends on the number of candidates that can be arrayed per surface—the more the better. Our method is suitable for automation and can be scaled up to a density of 25,000 spots per membrane, 35 which is comparable to the density of DNA chips used in gene expression studies. Other published protein arrays, such as chip-on-glass plates, 7 are able to accommodate about the same number of spots per surface (2413 spots for chip-on-glass plates). Moreover, with our technique, there is the possibility of screening additional membranes. The use of highly competent E. coli cells during peptide library construction provides enough material for the picking of thousands of colonies. Finally, E. coli is a fast and robust expression system with no posttranslational modifications in the expressed peptides. The use of microorganisms that do perform posttranslational modifications 36-38 could complicate efforts to map human antigens.
Apart from technical issues, the use of protein arrays is beneficial for clinical diagnostics and research. Three percent of the world’s population suffers from autoimmune diseases, 24 and one application of existing antigen arrays is to map the epitopes recognized by autoantibodies. Such mapping of autoantibodies is important in clinical diagnostics. For example, epidermolysis bullosa acquisita is characterized by autoantibodies against type VII collagen. 39 The method we described here is applicable to profiling the antibodies in sera of patients with autoimmune diseases. Its application to other diseases such as cancer or infectious diseases is also possible. There are observations that patients with cancer produce antibodies against tumor-specific antigens. 40-42 Our method would allow the construction of whole-proteome libraries for screening of new autoantibodies or for comprehensive, whole-proteome cross-reactivity validation of detection antibodies. 43 In infectious disease research and diagnostics, the mapping of antipathogen antibodies as well as the determination of antigens that elicit an immune response would be useful in drug discovery and give molecular insights into the immune response.
Besides diagnostics, the use of epitope mapping would also be beneficial to the field of antibody therapeutics. After vaccines, antibodies are the second largest class of drugs for human therapeutics. The development of potent antibody therapeutics involves the generation and optimization of antibodies that are used in several clinical settings such as oncology or infectious diseases. Thus, many antibodies are already in therapeutic use. 44 Most of the numerous antibodies that are entering clinical trials are derived from phage display libraries 45 or transgenic mice. 46 An example of a phage display–derived antibody for therapeutic use is adalimumab (Humira, Abbott Laboratories [Abbott Park, IL], a tumor necrosis factor [TNF]–specific monoclonal antibody); panitumumab (Abgenix, Inc. and Amgen Inc. [Thousand Oaks, CA]; an epidermal growth factor receptor-specific monoclonal antibody) is as example of a transgenic mouse-derived human antibody. Both antibodies act through receptor or ligand binding. Less widely used methods for the creation of human antibodies are ribosome, mRNA, and yeast display libraries. 45 The generation of antibody therapeutics proceeds iteratively. The steps are antigen selection, development of a therapeutic concept, antibody design, antibody generation followed by in vitro evaluation, and multiple tests and clinical trials in which the antibody is optimized until it has become an approved drug.
The second major step in creating antibody therapeutics is the optimization of the generated antibodies. Those antibodies have several factors that can be optimized to increase their therapeutic function. This includes antigen specificity and antigen binding affinity, effector functions, immunogenicity, and some other biological or biophysical properties. 47-49 The most widely studied property is the antigen binding affinity where several techniques for affinity maturation have been developed. 45 The binding affinity is denoted as the Kd value, which for antibodies in therapeutic use ranges from 0.1 to 32 nM. 44 Once a strong antibody is identified, increasing its antigen specificity would be another focus for improvement. For this, knowing the specific epitope of the antibody would be of great interest. Therefore, the method described here represents a cost-effective step in the cost-intensive generation of antibody therapeutics.
Last, because each found epitope is expressed in a bacterial expression vector and fused with an affinity tag, the fusion protein can be used for affinity purification of epitope-specific antibodies. In this way, polyclonal antibodies out of sera could be purified and further examined.