
Abstract
Historically, traditional screening for ligands has been optimized to detect standard orthosteric agonists and antagonists. However, with increasing emphasis on cellular functional screens, more allosteric ligands are being discovered as potential drugs. In addition, there are theoretical reasons (increased selectivity, better control of physiological systems, separate control of affinity and efficacy) allosteric ligands may be preferred therapeutic chemical targets. These factors may make it desirable to design high-throughput screens to specifically detect functionally allosteric ligands. This article discusses the unique features of allosteric ligands as drugs as well as the special conditions that should be considered to optimize a high-throughput screen toward the detection of allosteric drugs. Finally, the likelihood of detecting allosteric ligands that have direct effects on cells (either conventional agonism or functionally selective effects) is discussed as well as the optimization of detection of such ligands in screening assays.
Keywords
Introduction
T
There are 2 basic mechanisms by which a molecule can alter the behavior of a 7TM receptor toward other molecules: ortho-steric or allosteric interaction. Orthosteric interaction refers to a steric competition between ligands as they compete for a common binding site on the receptor with an agonist or radioligand (defined as the receptor probe). In orthosteric systems, there is never a protein species with both orthosteric ligand and probe bound at any one instant. Therefore, the nature of the probe cannot influence the effect of the ligand. In this sense, orthosteric systems are preemptive in that the receptor response is to either one or the other ligand in the system. 3 Allosteric ligands bind to their own site on the receptor and affect endogenous ligand receptor effects through a conformational change in the receptor. Such systems are permissive in that there are receptor protein species with both ligand and receptor probe bound simultaneously. 3 Under these conditions, the nature of the probe can influence the effect of the ligand. Allosteric systems are much more versatile than orthosteric systems in that modulator ligands can augment, block, or potentiate the effects of probes, and this effect can change with the nature of the probe. There is an increasing awareness of allosteric ligands both from the point of view of appreciation of what these types of ligands can do therapeutically 4 and also their increasing numbers as hits in screening assays. This latter phenomenon probably is due to the increasing utilization of functional screening assays. 5 Thus, although high-throughput binding assays are biased toward the detection of ligands that influence the interaction of the receptor with a single receptor probe (orthosteric interaction or limited to allosteric alterations in the affinity of the probe), functional assays have the capability to detect ligands that allosterically alter the affinity and/or efficacy for any species that interacts with the receptor, including cytosolic signaling proteins. This casts a wider net of detection for receptor ligands. This article focuses on the optimal conditions required for the detection of allosteric ligands. As a preface to this discussion, it is useful to define what 7TM receptor interactions are contained within the set of behaviors referred to as allosteric.
What is Receptor Allostery?
Protein allosterism is defined as the effect on a protein (receptor) produced by the simultaneous interaction with 2 molecules. 7TM receptors are nature’s prototype protein as they are designed to bind small molecules in one region and change their conformation in response to affect the interaction of the receptor with another ligand in another region. If that other ligand is a cytosolic signaling protein (e.g., G-proteins, β-arrestin), then the allosteric ligand can cause cellular response. These same mechanisms can be channeled to cause one molecule (defined as the allosteric modulator) to change the behavior of the receptor toward another ligand; this molecule will be referred to as the “guest” (note that guests whose presence can be detected on the receptor can be probes). It should be noted that, unlike orthosteric interaction of molecules with the receptor (where the molecule binds to the endogenous agonist site), there is no limitation on the effect of an allosteric modulator. A useful way to view an allosteric interaction is to consider the allosterically modified receptor as a new receptor type with potentially different affinities and efficacies for all interactants (guests).
Two important characteristics of allosterism should be considered; the first is that allosterism involves relationships between bodies, and thus allosteric effects must be defined in terms of the modulator ([B]) and the guest ligand on the receptor (allosteric effects must be defined for distinct modulator/receptor/guest complexes). The second feature of allosterism is that it is vectorial in nature. Thus, a modulator can increase or decrease guest affinity and/or efficacy. Moreover, there are no rules governing how these effects are linked (i.e., affinity may be increased while efficacy is decreased, etc.). This will be discussed in more detail in the following section.
To effectively quantify allosteric effects, it is essential to have a receptor model to describe, in numbers, all possible allosteric modification of a probe concentration-response curve. This enables identification of parameters that can be used to quantify allosteric properties of modulators for medicinal chemists as they explore structure-activity relationships. A simple and concise model for functional allosterism defines modification, by an allosteric modulator [B], of a response to an agonist [A] with a combination of the Ehlert model for allosterism 6 and the Black/Leff operational model for receptor function. 7 This model predicts the effects of probe [A] in the presence of an allosteric modulator [B] as 8-10 (derived in the appendix)
where Emax is the maximal response returned by the system, and the equilibrium dissociation constants for the cobinding ligands [A] and [B] are KA and KB, respectively. The factor τ represents the pharmacological efficacy of the agonist in the particular functional system, α is the change in the affinity of the cobinding ligand (agonist) when the modulator is bound, and β is the change in efficacy of the cobinding ligand with modulator bound. The model has the capability of describing virtually any change in the concentration-response curve to a receptor probe and converting the changes into numbers. In this way, allosteric effects can be quantified through the magnitude(s) and vectorial properties of α and β. It is worth considering these numbers as descriptors of allosteric change in a receptor to define 3 basic properties of allosteric modulators.
Basic Features of Allosterism
The permissive nature of allosteric systems gives them 3 unique pharmacological behaviors. These are as follows.
Saturation of effect
Because allosteric modulators bind to their own site on the receptor, their effects will be saturable, and no further effect of the modulator will be observed when the allosteric site is saturated. This is in contrast to orthosteric systems, which theoretically can show competitive kinetics for as long as varying concentrations of probe and antagonist are added to the system. The saturation of effect property allows allosteric modulators to change but not completely eliminate the effect(s) of probes. For example, the ligand UCB35625 reduces but does not completely eliminate the binding of 125I-CCL3 to the CCR1 receptor. 11 Thus, a modulator with an α value of 0.2 will produce a 5-fold reduction in the affinity of a receptor probe. Therefore, depending on the concentration of probe in the system, the inhibitory effects of such a modulator may be manifest as incomplete antagonism or “partial antagonism.” Figure 1 shows the effects of such a modulator, specifically a compound that produces a 5-fold reduction in the affinity of the receptor for the probe ligand with no change in the probe efficacy. The inverted sigmoidal curve shows the effects of the modulator on a concentration of agonist that produces 80% maximal response (an EC80 concentration). It can be seen that the maximal effect of the modulator is to reduce the agonist effect to 30% maximum; no amount of modulator will produce further antagonism. Should this effect occur in vivo, it would reduce but not completely block the function of the receptor.
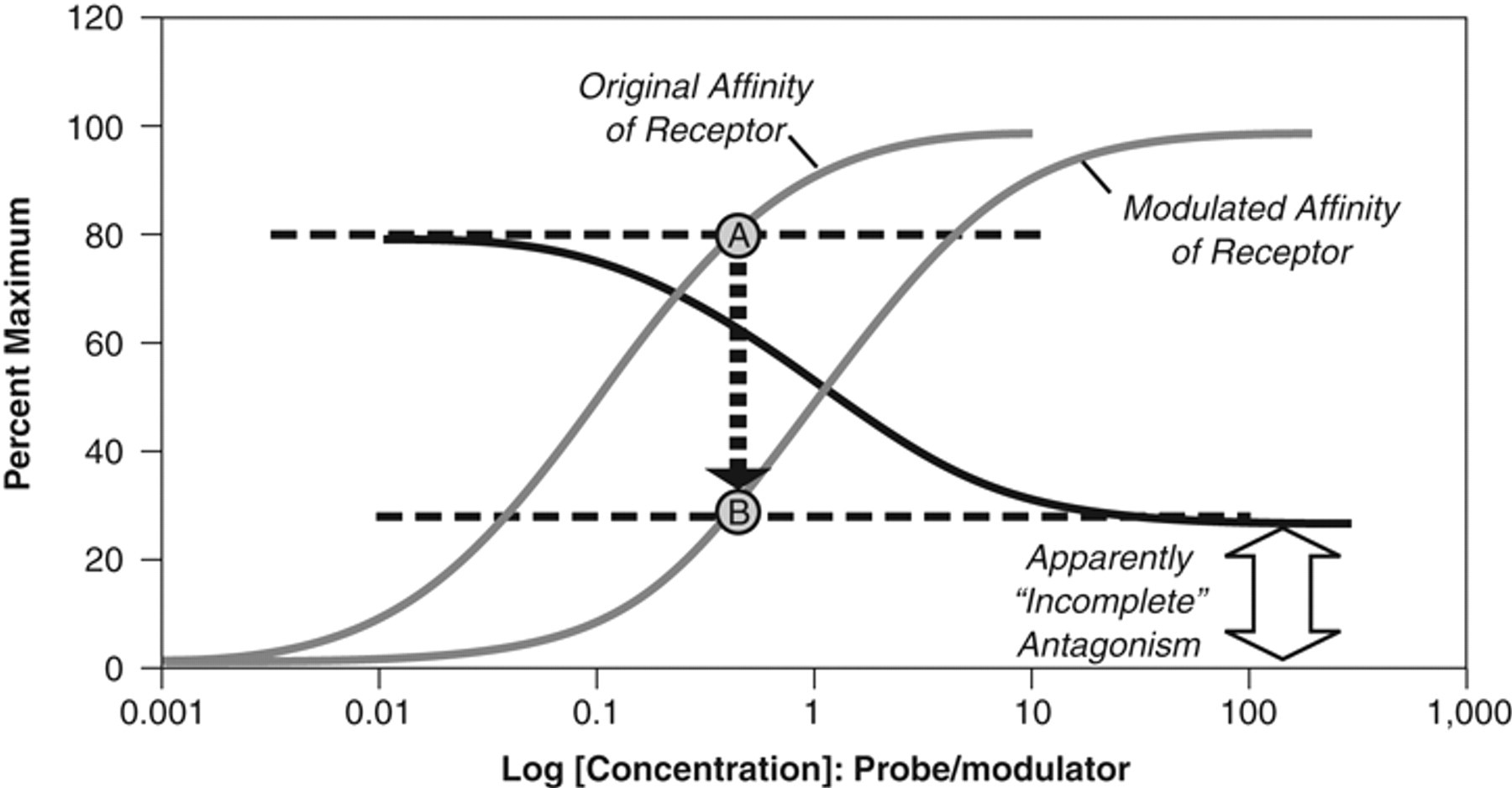
Surmountable antagonism by an allosteric modulator antagonist with equation (1) values of α = 0.2, β = 1, τ = 9 (abscissae = [A]/KA ratios). The modulator produces a 5-fold shift of the agonist concentration-response curve to the right (curves drawn in gray). An inverse sigmoidal inhibition curve would be produced by the modulator in blocking an EC80 concentration of the agonist (point A). The modulator would produce a maximal diminution of response to point B coinciding with the maximal decrease in affinity (5-fold) produced. The response to the agonist would decrease to a level producing a residual 30% response; this would not be eliminated by the modulator at any concentration. This has erroneously been referred to in the literature as to be partial antagonism but rather is the resetting of the affinity of the protein for the ligand.
The effects shown in Figure 1 underscore another feature of allosteric antagonists—namely, the unpredictable relationship between their ordinate effects on functional response and their potency; this is in contrast to orthosteric antagonists. In the latter case, once a concentration of antagonist is added to the system that produces a threshold antagonism, it is predicted that a 10-fold greater concentration will produce 50% inhibition, and 100-fold greater concentrations will nearly completely block the response. In this sense, there is a defined relationship between the ordinate response scale and the concentrations of antagonist present in the system. In contrast, for allosteric modulator antagonists, the relationship between the occupancy of the allosteric site and the resulting effect on the agonist response is defined by the magnitude(s) of α and β. For example, in the case shown in Figure 2 , 10 times the threshold concentration of modulator produces 30% inhibition, and a 100-fold increase produces a maximum inhibition of only 60%. This effectively produces a disconnection between the vertical readout of response inhibition as used by “1-shot screening” formats and compound activity. This, in turn, may lead to a different choice of concentration for screening, using interference of the effects of a single concentration of receptor probe (i.e., agonist or radioligand). Figure 2 shows the antagonism of an EC80 concentration of agonist as blocked by an allosteric modulator and an orthosteric antagonist. It can be seen that the modulator is potent (low IC50) with a modest maximal effect. Testing a low concentration would detect this antagonist (for the particular antagonist shown in Fig 2 : Zone I) but not detect the effects of the orthosteric antagonist shown. Testing at a higher concentration (Zone II) would detect the orthosteric antagonist but show little increase in the effect of the allosteric modulator. In effect, allosterism can differentiate vertical-scale ordinate effects on agonist activity from horizontal location parameters denoting allosteric potency. If an allosteric modulator is sought from the outset of the screen, it might be more beneficial to screen at lower concentrations as this may reduce nonspecific effects (i.e., higher concentrations may not necessarily translate to more hits as they would in orthosteric screens).
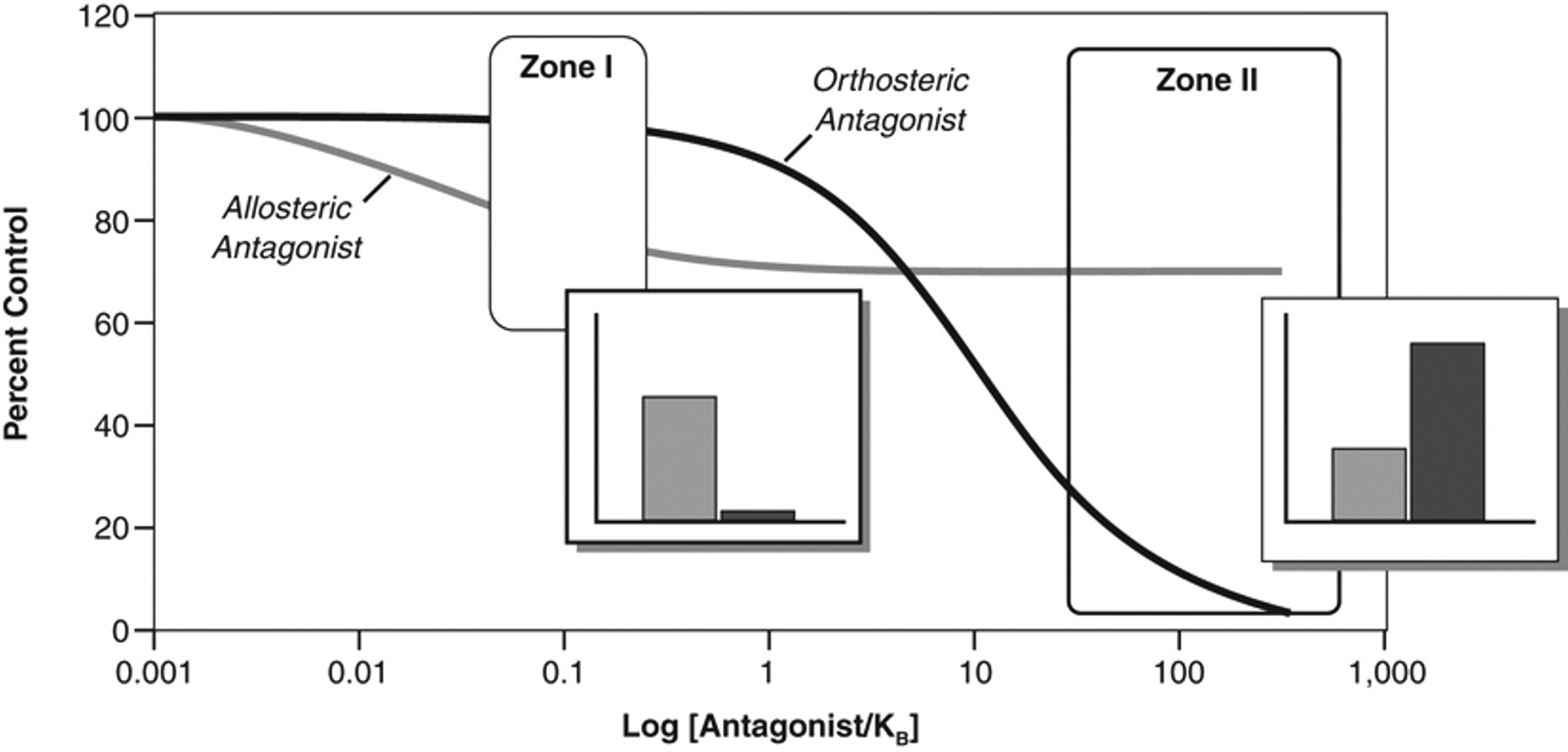
Inhibition curves (blockade of an EC80 concentration of an agonist) produced by 2 hypothetical antagonists; one produces orthosteric blockade (black line) and one an allosteric diminution of agonist affinity (gray line). Receptor probe concentration set at [A]/KA = 10; odulator α = 2, β = 1. Zones I and II show regions of antagonist concentration chosen for single-shot antagonism. Zone I is a lower concentration ([B]/KB = 0.1). The inset bar graph shows the observed antagonism (ordinate axis is % inhibition). It can be seen that the orthosteric antagonist produces no significant effect while the allosteric antagonist produces a measurable signal. Zone II is a higher concentration of antagonist ([B]/KB = 100) where it can be seen that the allosteric signal is unchanged while the orthosteric effect is visible.
Probe dependence
There is no a priori reason to think that allosteric changes toward a set of guest interactants will be uniform (i.e., if the affinity of one muscarinic agonist is reduced by a factor of 10 by an allosteric modulator, it should not be assumed that the affinity of all muscarinic agonists also will be reduced by a factor of 10). This effect, referred to as probe dependence, 12 is clearly shown by the allosteric modulator alcuronium, which reduces the affinity of muscarinic radiolabeled antagonist [3H]-methyl-QNB but increases the affinity to the radioactive antagonist [3H]-atropine. 13 Because of probe dependence, it is important that the physiologically relevant agonist be used in the screening process if possible. This can be in conflict with practical aspects of screening where stability and ease of handling of probe molecules can be important. For example, there is theoretical rationale for the potentiation of signals mediated by the cholinergic nervous system in the brain for the treatment of Alzheimer’s disease. 14-18 In light of this hypothesis, an allosteric potentiator of neuronal cholinergic effect could be therapeutically useful. However, acetylcholine, the physiologically relevant neurotransmitter, is unstable and difficult to use in high-throughput screening (HTS), raising the question of whether a more stable cholinergic agonist (e.g., pilocarpine) should be used in an allosteric screen for potentiation of cholinergic agonism. The danger of doing so is the potential for probe dependence to detect potentiation of the surrogate ligand that will not transfer to potentiation of the natural neurotransmitter acetylcholine. In fact, there are examples of dissimulations in potentiator activity with different muscarinic agonists. Although allosteric potentiation of the effects of surrogate muscarinic agonists such as pilocarpine and 5-thio-TZTP are produced by alcuronium, brucine, and strychnine, the effects on the physiologically relevant neurotransmitter acetylcholine are opposite (antagonism) and therefore of no utility in Alzheimer’s disease. 19
Probe dependence can be misleading in the screening process, but it also can be therapeutically advantageous as in the inhibition of HIV-1 virus infection through allosteric blockade of the CCR5 receptor. In this case, data show that a functioning chemokine system through chemokine CCL3L1 activation of CCR5 can yield protective effects in AIDS, as inferred by the correlation between survival after HIV-1 infection and gene copy number for CCL3L1. 20 It is thought that chemokines are beneficial in AIDS because they cause internalization of CCR5, thereby removing the site for infection (the CCR5 receptor) from the cell surface. In agreement with this idea, it has been shown that there is a reduction in cell surface CCR5 levels associated with CCL3L1 gene copy number. 21 This suggests that an allosteric blocker that prevented binding of HIV-1 but otherwise preserved CCL3L1 function would have increased therapeutic efficacy. 22 Such a functional selectivity (blockade of HIV-1 binding with preservation of chemokine function via CCR5) can only be achieved with an allosteric modulator. In fact, it has been shown that 6 CCR5 allosteric modulators have a relative range of selective potency for HIV versus CCL3L1 internalization of almost 500, suggesting that selective probe dependence through allosteric modulation of the CCR5 receptor is possible. 22
Selective effects on affinity and efficacy
In accordance with the notion that an allosteric modulator can stabilize a unique receptor conformation, both the affinity and/or efficacy of guest probe molecules can be different for a modulated receptor as compared to what they were on the wild-type receptor. For example, the CCR5 allosteric modulator aplaviroc has little effect on the affinity of the receptor for the chemokine CCL5 (as measured by binding of the radioactive chemokine 125I-CCL5) but does prevent CCL5 from producing pharmacological response (see Fig. 3 ). 23,24
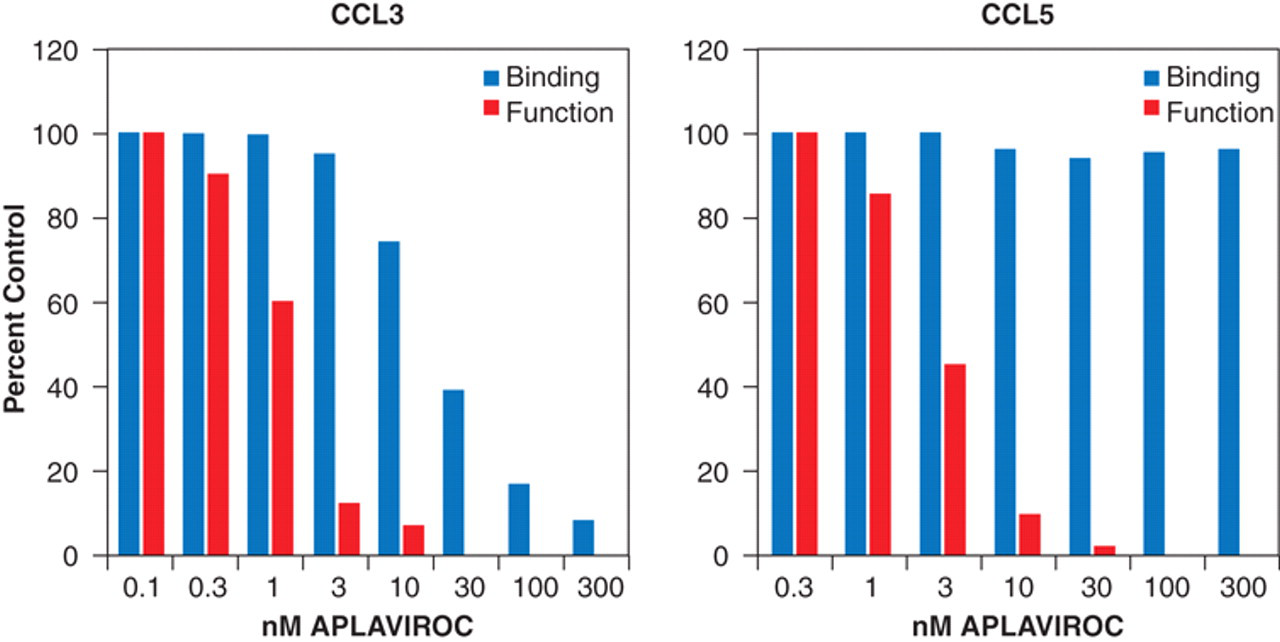
Effect of the CCR5 allosteric inhibitor of HIV-1 entry aplaviroc on CCR5 receptor binding (light gray bars) and CCR5-mediated calcium response function (dark gray bars) of the chemokines CCL3 and CCL5. It can be seen that although aplaviroc blocks both binding and function of CCL3, only the functional effects of CCL5 are blocked. Data from Watson et al. 23
Reductions in α and/or β will result in reduced agonist activity and antagonism. Specifically, reducing α (reduced affinity) with no change in agonist efficacy (or even increased efficacy) will result in dextral displacement of agonist concentration-response curves. This profile can resemble, but should not be confused with, simple competitive antagonism. Similarly, a reduction in β (blockade of agonist efficacy) will produce depression of the maximal response to agonists, although this can be offset by receptor reserve in the system. In systems of high receptor reserve, a modulator that blocks efficacy will produce dextral displacement with little diminution of maximum. However, at higher levels of receptor blockade, diminution of maximal responses will be observed. In systems where there is no receptor reserve, little dextral displacement and depression of maximal response will be seen.
For allosteric antagonists (β = 0), opposing effects on affinity and efficacy can result in some interesting profiles of activity. For example, if a modulator increases affinity for the agonist but decreases efficacy, then a condition where the potency of the antagonist is linked to the degree of activation of the system can result. Specifically, the reciprocal nature of the modulator-probe interaction predicts that, for modulators that increase the affinity of the receptor for the probe, the presence of a probe reciprocally will increase the affinity of the receptor for the modulator. This being the case, a chemical context whereby the assay is conducted in the presence of some concentration of probe will increase the sensitivity of the receptor to these types of modulators. This is evident from equation (1), which predicts that the IC50 (molar concentration producing 50% inhibition of a defined agonist response) for an allosteric antagonist is given by
Equation (2) defines a relationship between α and the observed affinity of a modulator as quantified by the IC50. This relationship shows that if an allosteric modulator reduces the affinity of the agonist (α << 1), the relationship between the IC50 and the KB reduces to the relationship identical to that operative for simple competitive antagonists. 25 However, if the modulator increases the affinity of the receptor for the agonist (α > 1), then higher concentrations of agonist will actually increase the potency of the antagonist modulator ((IC50/KB) > 1). Thus, the antagonist will adjust its potency according to the level of activation of the system. In essence, this would be the ideal antagonist because it would have minimal effects until the system is activated. Such a profile is seen with the NMDA receptor allosteric modulator ifenprodil, which increases the affinity but decreases the efficacy of the agonist NMDA. 26 Figure 4 shows the inverse relationship between the level of agonism in the system and the IC50 for ifenprodil (i.e., note the increase in potency of ifenprodil as a blocker in the presence of 100 μM NMDA as opposed to 10 μM NMDA).
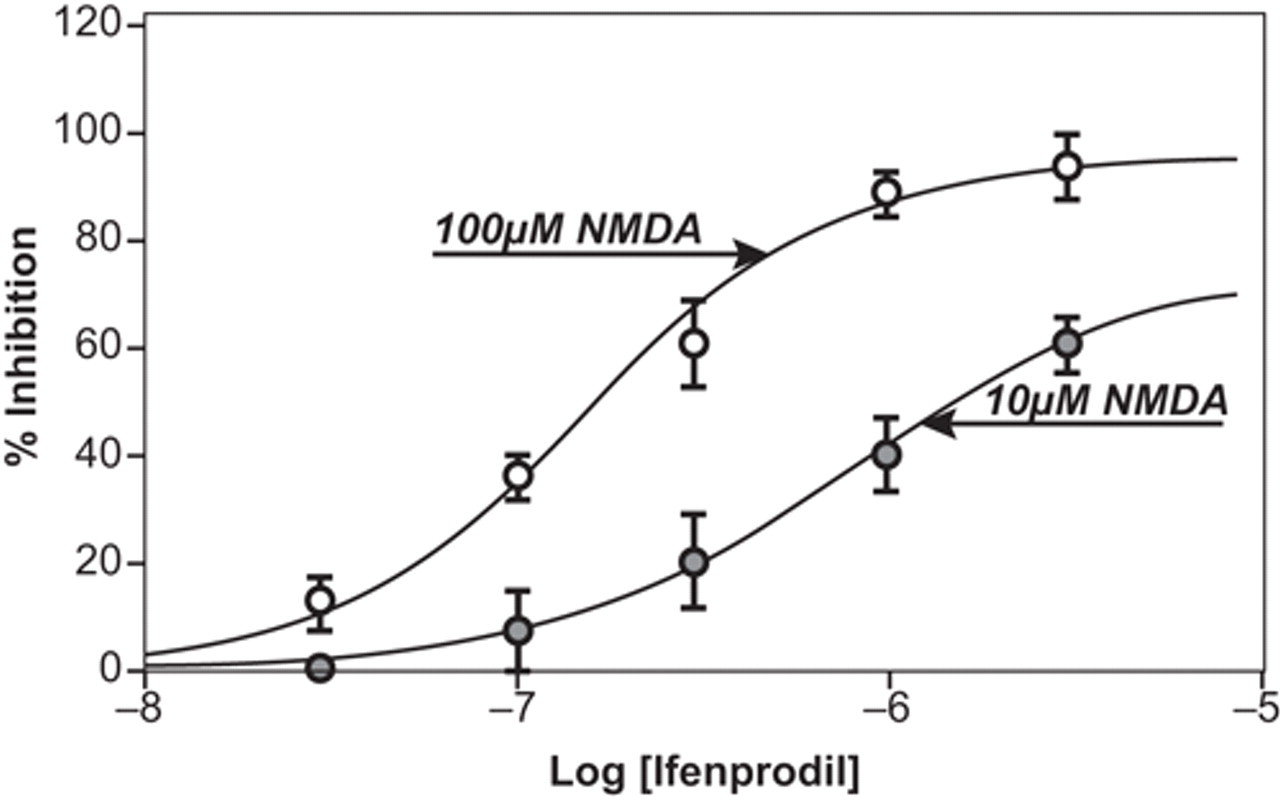
Blockade of the NMDA receptor-mediated effects of 2 concentrations of NMDA by the allosteric modulator ifenprodil. It can be seen that ifenprodil is more potent in blocking the higher concentration (100 μM) of NMDA, showing a relationship between potency and intensity of activity of the system. Data redrawn from Kew et al. 26
Another aspect of allosteric antagonism is the potential to differentially alter the efficacy of an agonist for different signaling pathways—in effect, to impose functional selectivity on endogenous agonists. For example, the allosteric modulator Nα-tosyltryptophan causes the CRTH2 receptor natural agonist prostaglandin D2 to change its activation profile from that of activating Gi and β-arrestin to sole activation of Gi-protein (with no concomitant β-arrestin interaction). 27 Similarly, the natural NK2 receptor agonist neurokinin A activates Gs and Gq, whereas the allosteric modulator LP1805 changes this pattern to one of enhanced Gq and blockade of Gs activation. 28
Finally, it is worth considering a causal link between kinetics of action and allosterism. It has been noted that the rate of onset and dissociation of allosteric enzyme inhibitors can be much longer than for orthosteric ligands. In the case of some p38 mitogen-activated protein (MAP) kinase inhibitors, the rate of onset of allosteric inhibitors is 500 times slower than for orthosteric antagonists. 29 The mechanism for these longer onsets has been postulated to relate to the small amount of time an allosteric binding site may be open in the conformational lifetime of the protein, as opposed to the time an orthosteric site must remain open to accommodate the endogenous substrate. In practical terms, these long onset times bring with them a requirement for long preincubation times in assays and screens aimed at determining properties of allosteric compounds. On the other hand, long onset times also usually mean long offset times (slow rates of dissociation), leading to better target coverage. For example, allosteric inhibitors of HIV-1 entry have half-times for dissociation from the CCR5 receptor ranging from 80 to >300 h. 23
Positive Allosteric Modulators
Allosteric modification of receptor function can result in potentiation of agonist response (for examples, see references 30-34 ); ligands that produce this effect are referred to as positive allosteric modulators or PAMs. A positive allosteric modulator could potentiate agonist response either through effects on affinity (α > 1) or efficacy (β > 1); equation (1) can be used to predict positive modulation of response for different types of potentiators. Shown in Figure 5A is a modulator with α = 5 at a concentration of [B]/KB = 20. It can be seen that the concentration-response curve to the agonist is shifted to the left. A plot of the PAM-induced increase in response at any defined level of control response yields a bell-shaped curve shown to the right of the concentration-response curves. This curve depicts the increased response observed with the PAM at various levels of control response. For the example shown in Figure 5B , this analysis is continued for a modulator that increases the affinity of the agonist (curves shown for α = 2, 3, 5, 10, and 20) for concentrations of modulator [B]/KB = 0.1, 0.33, 1, 3.3, 10, and 30. For a screening assay run at 10 μM, it might be assumed that this would correspond to [B]/KB values of 0.1 to 10. In general, it can be seen that the maximal sensitivity to the modulator occurs between control response levels of 0.2 and 0.4, suggesting that the optimal levels for receptor probe (agonist) presence in the assay (referred to as the chemical context) would be between the EC20 and EC40 values. In view of the need to preserve an optimal window for response visualization, this would suggest that the EC20 would be optimal. A virtually identical profile is predicted for a modulator that increases the efficacy of the agonist (β > 1 or mixed increases in α and β; data not shown). In general, theoretical analyses indicate that the EC20 is the optimal chemical context for detection of positive modulator activity.
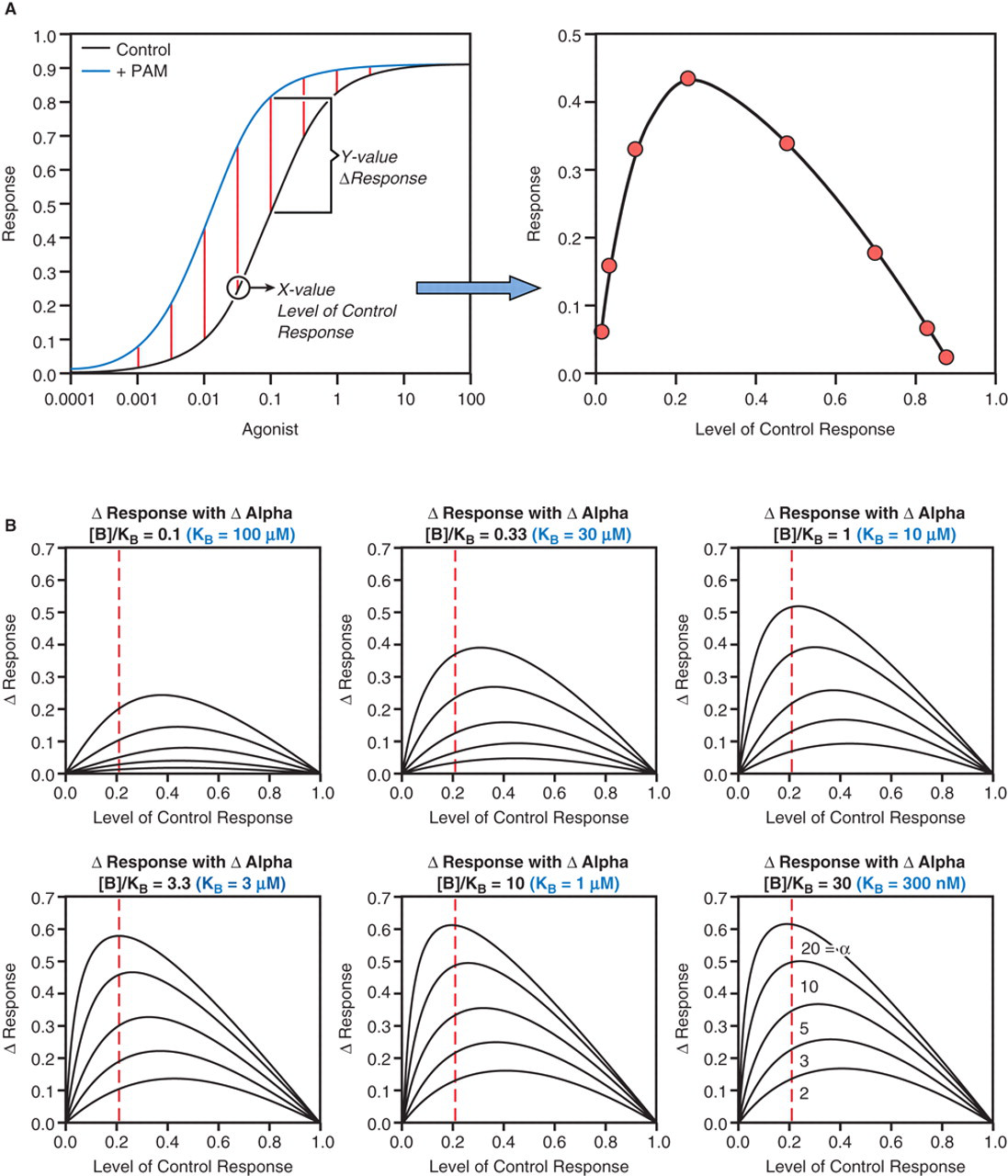
(
Allosteric Agonists
There is no theoretical reason an allosteric change in the receptor would not alter the interaction of the receptor with ligands and cytosolic signaling proteins; this latter effect could stabilize an active state of the receptor to produce agonism. The model describing these effects 35,36 yields the following relationship for the activity of a probe agonist in the presence of an allosteric agonist modulator:
where φ refers to the ratio of intrinsic efficacies of the modulator agonist B and probe agonist A (φ = τB/τA). There are 3 properties of allosteric agonists of potential relevance to their detection and therapeutic use. The first is that the agonism produced by these ligands may be insensitive to conventional orthosteric antagonists. For example, the conventional orthosteric receptor antagonist QNB is ineffective in blocking the effects of the muscarinic allosteric agonist alcuronium. 37 This may raise questions regarding target validation—that is, whether the allosteric agonist produces agonism through interaction with the specific target of interest. Second, it is important to note the effect that an allosteric agonist may have on the endogenous agonist tone in the body. Although a conventional orthosteric partial agonist will block the effects of the endogenous agonist, an allosteric agonist may not and, in fact, may even potentiate basal tone. This may cause fundamentally different behaviors in vivo from the effects seen with a conventional partial agonist and also can furnish ways to differentiate allosteric agonism from orthosteric agonism even at the screening stage. Figure 6 shows the effects of an orthosteric partial agonist and an allosteric partial agonist on a concentration of probe agonist that produces 80% maximal response (EC80). It can be seen that although the orthosteric agonist produces direct agonism (10%), it blocks the EC80 response. In contrast, the allosteric agonist does not interfere with the EC80 response. This profile will be true for allosteric agonists that have α and β values >1; for allosteric agonists that have a negative allosteric effect on the probe agonist, the profile will resemble that of an orthosteric partial agonist. 35
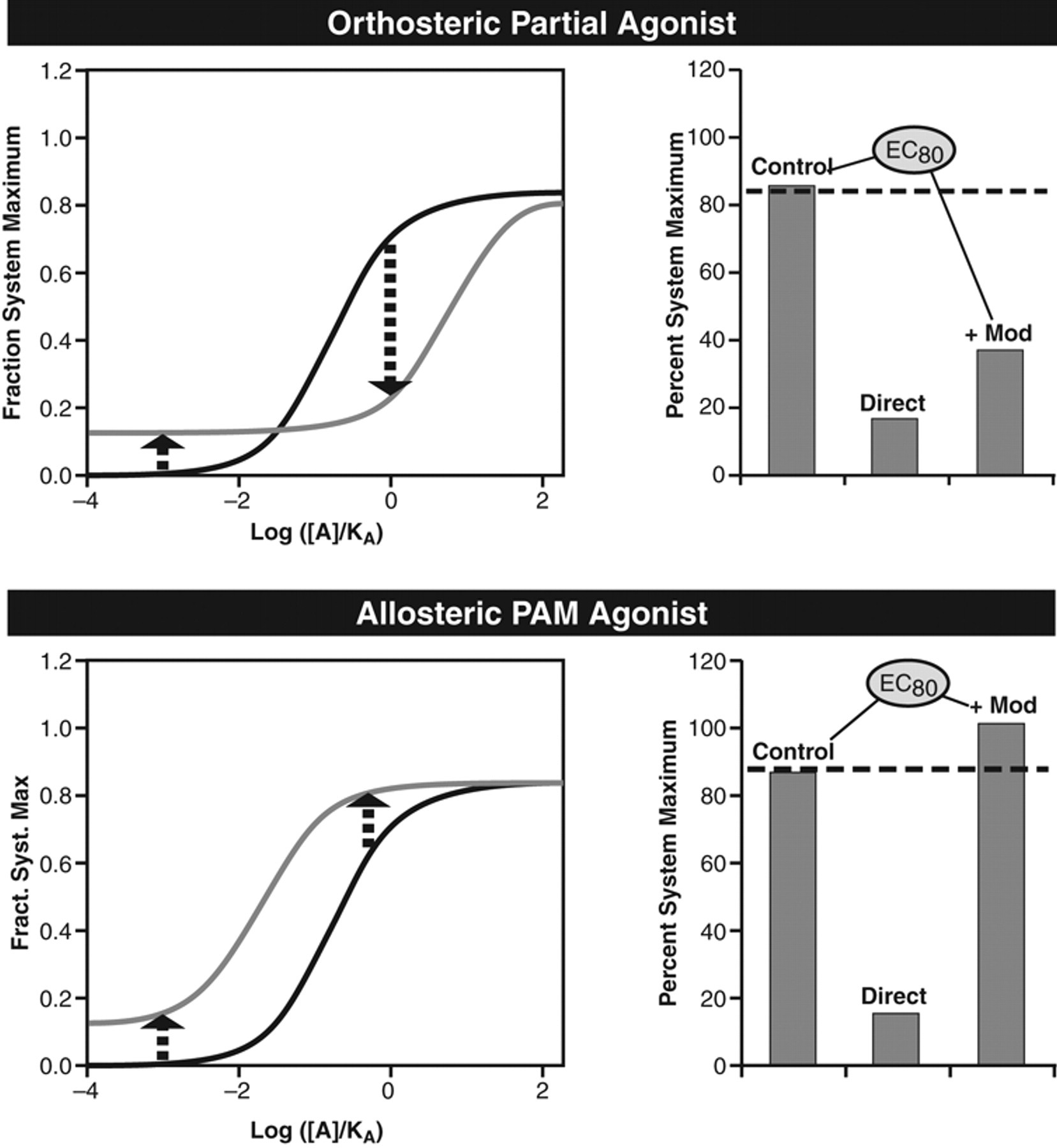
Direct effects of 2 kinds of partial agonist and their effects on an EC80 concentration of probe agonist. Top panel shows the effect of a conventional orthosteric partial agonist on the concentration-response curve to the probe agonist (elevated baseline and dextral displacement). Histograms on the right show a hypothetical EC80 response to the probe agonist (first bar), direct effect of the partial agonist (second bar), and effect of the partial agonist on the EC80 concentration of the probe agonist (third bar). It can be seen that the partial agonist reduces the EC80 effect of the probe agonist. Bottom panel shows the effect of an allosteric partial agonist on the concentration-response curve to the probe agonist (elevated baseline and no displacement or sinistral displacement). As with the top panel, histogram on the right shows a hypothetical EC80 response to the probe agonist (first bar), direct effect of the partial agonist (second bar), and effect of the partial agonist on the EC80 concentration of the probe agonist (third bar). It can be seen that, in this case, the partial agonist does not reduce the EC80 effect of the probe agonist. Redrawn from Kenakin. 35
Finally, allosteric agonists may have different signaling characteristics from the natural endogenous agonist(s) for the receptor. Recent data in the literature have confirmed that agonists may stabilize selective receptor active states to yield functionally selective agonism. 38-42 Because 7TM receptors may interact with a range of different cytosolic reactants, it should not be assumed that activation of the receptor by an allosteric agonist will activate the same range of signaling molecules as the endogenous agonist. For example, selective signaling by allosteric agonists has been reported (effects of AC-42 on muscarinic M1 receptors 43 ). Therefore, different agonist pathways should be considered for allosteric agonists. A major signaling pathway that can be kinetically differentiated from G-protein activation is activation of ERKinase via β-arrestin. 44-46 Figure 7 shows the kinetics of response production by angiotensin, where it can be seen that the β-arrestin-mediated response has a longer onset time and is more prolonged than the G-protein signal. 47 This figure shows how the temporal window of detection of a screening assay can bias detection from G-protein to β-arrestin activation. Therefore, with the increased potential of allosteric agonists to activate alternative signaling pathways, the window for agonist detection should be considered an important variable.
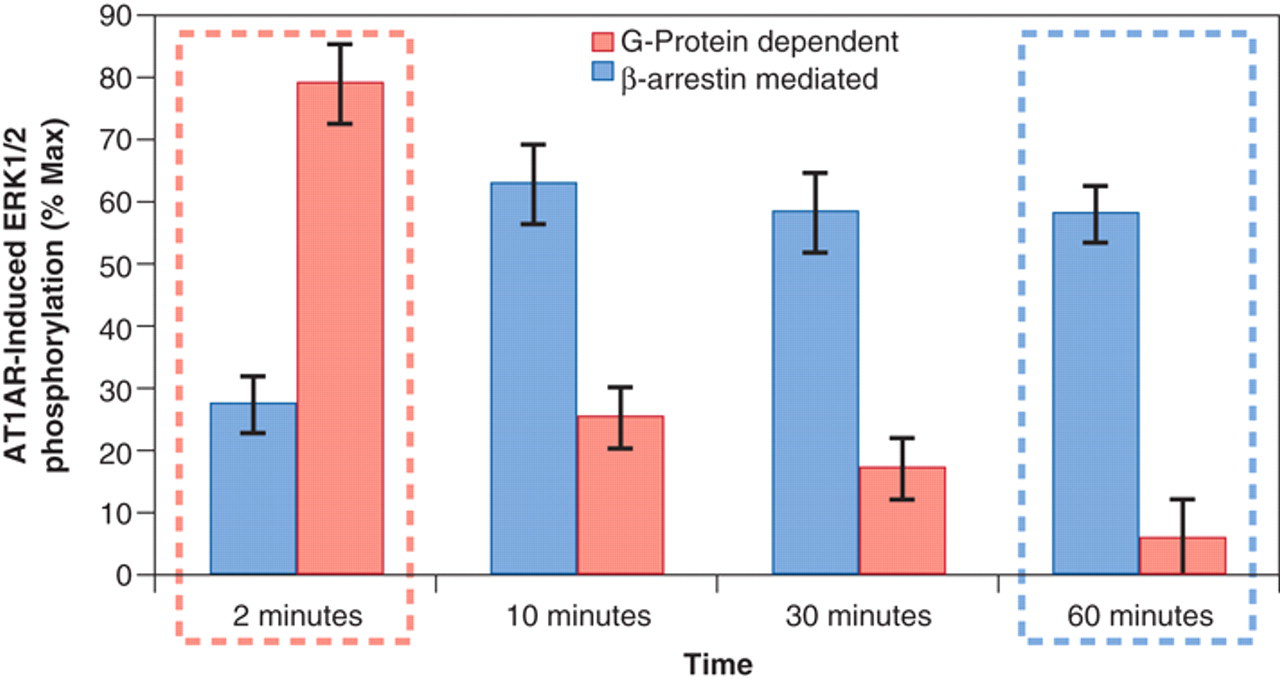
Responses to angiotensin-1 taken at various time points from various parts of the signaling cascade. The bars on the left show G-protein-mediated responses, and the bars on the right show β-arrestin (G-protein-independent) mediated extracellular signal-regulated kinase (ERK) activation. It can be seen that although a powerful G-protein activation can be seen at 2 min, little remains at 60 min. In contrast, a powerful β-arrestin-mediated response seen at 60 min is of lower magnitude at 2 min. Drawn from Ahn et al. 47
Chemical Context
In light of the interactive and permissive nature of allosterism, the chemical context (presence of a ligand in the screening milieu) of screens designed to detect allosteric molecules may need to be optimized. Clearly, for all antagonists, orthosteric or allosteric, there needs to be a probe agonist in the system to detect antagonism. Usually, the concentration of probe agonist needs to be large enough to produce an adequate window of detection for antagonism but not too large so as to produce loss of sensitivity due to competitive effects. 25 In the case of allosteric antagonists, where effects often are noncompetitive, the concern of lost sensitivity with tracer agonist concentration in the medium is less of an issue (unless α << 1). Also, as demonstrated by equation (3) and exemplified by ifenprodil (see Fig. 4 ), if an allosteric antagonist increases agonist affinity but decreases efficacy, presence of a tracer agonist in high concentration can actually enhance detection by increasing the receptor affinity for the modulator.
A situation where chemical context is critical is in the detection of PAMs. A PAM with no intrinsic efficacy of its own (φ = 0; equation (3)) will produce a visible effect only in the presence of the endogenous agonist (see Fig. 5 ). As seen in Figure 6B , the presence of an EC20 concentration of agonist is optimal for detection of positive modulator activity. Figure 8 shows 1-shot screening results for 500 virtual ligands with randomly assigned values for affinity (KA), α (effect on endogenous affinity; for simplicity, it is assumed that β = 1), and φ (intrinsic activity as agonists in their own right). As expected, in the absence of chemical context, only the direct agonists are detected. In the presence of a tracer amount of endogenous agonist, a larger sample of compounds is seen.
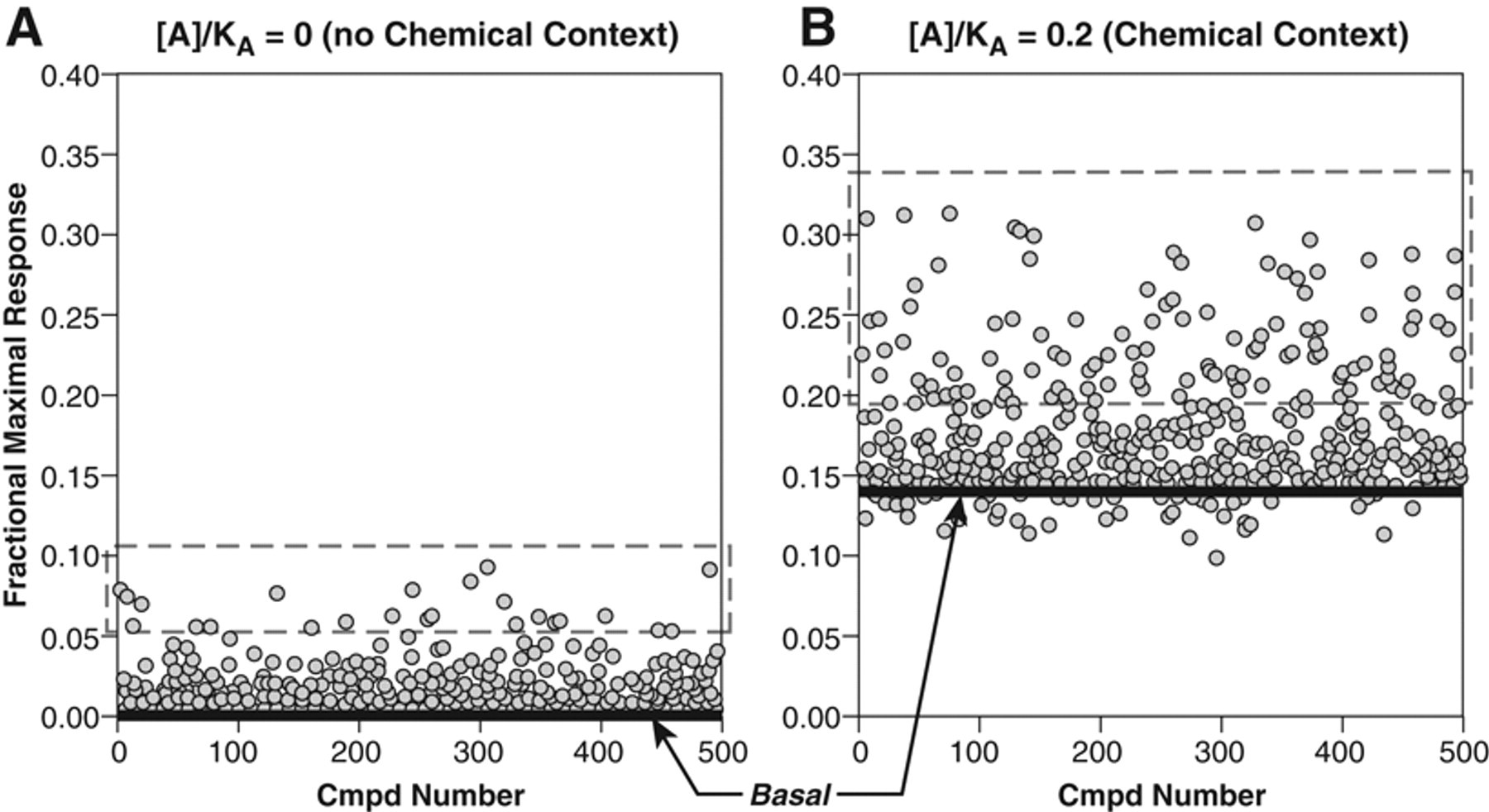
Hypothetical 1-shot screening data for 500 ligands with randomly assigned values
of affinity KA, α, and φ (see equation (3)). Compounds with φ
> 0 represent direct agonists, whereas those with α < 1 and allosteric
antagonists and α > 1 are PAMs. Panel
Conclusions
Pharmacological data show that the permissive nature of allosteric ligands gives them behaviors in pharmacological systems different from those of conventional orthosteric ligands. Similarly, there are conditions for finding such ligands that differ from those optimal for standard screening assays for conventional agonists and antagonists. For example, allosteric probe dependence dictates that the natural endogenous ligand be used for cobinding in the assay as opposed to a surrogate ligand optimized for robust activity under screening conditions. Another difference is the introduction of chemical context into the screen, specifically for PAMs where an EC20 level of stimulation allows optimal assay sensitivity. Finally, there should be an expectation of direct agonist or inverse agonist effects for allosteric ligands because the stabilization of a receptor conformation known to alter endogenous agonist effects could well alter the interaction of the same receptor with cytosolic signaling molecules. Such direct effects may not necessarily result in conventional second-messenger agonism but may, instead, relate to other 7TM receptor behaviors (i.e., β-arrestin interaction, receptor internalization).