
Abstract
Protein stabilization upon ligand binding has frequently been used to identify ligands for soluble proteins. Methods such as differential scanning fluorimetry (DSF) and differential static light scattering (DSLS) have been employed in the 384-well format and have been useful in identifying ligands that promote crystallization and 3D structure determination of proteins. However, finding a generic method that is applicable to membrane proteins has been a challenge as the high hydrophobicity of membrane proteins and the presence of detergents essential for their solubilization interfere with fluorescence-based detections. Here the authors used MsbA (an adenosine triphosphate binding cassette transporter), CorA (a Mg++ channel), and CpxA (a histidine kinase) as model proteins and show that DSLS is not sensitive to the presence of detergents or protein hydrophobicity and can be used to monitor thermodenaturation of membrane proteins, assess their stability, and detect ligand binding in a 384-well format.
Introduction
I
Experimental assessment of protein stability has previously been employed to identify ligands and optimum buffer conditions by a variety of methods. 4,6-8 Differential scanning fluorimetry (DSF) and differential static light scattering (DSLS) are 2 methods that recently have been used to screen soluble proteins against libraries of compounds in the 384-well format. However, the presence of detergent micelles adds complexity to the preparation of membrane proteins, limiting the application of the fluorescence-based methods such as DSF, causing high background and poor signal-to-noise ratios. Some attempts have been made to measure the stability of membrane proteins using DSF. 7,9,10 However, hydrophobic dyes such as SYPRO-orange used in DSF partition into detergent micelle and protein-detergent complexes and cause considerable increase in fluorescence background. Similarly, interaction of reporter dyes with solvent-accessible hydrophobic areas within some folded soluble or membrane proteins can cause a significant increase in the background. Low signal-to-noise ratios usually necessitate tedious data manipulation and background subtractions. In addition, in some cases, the presence of a reporter dye may affect stability and folding state of the protein. 11,12 Chemical reactivity of the cysteine residues embedded in the interior of proteins has also been used to detect protein unfolding. In this method, thiol-specific probes such as CPM (N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide) were used to monitor membrane protein unfolding by detecting the exposed thiol groups during protein denaturation. 13 However, this method has significant limitations; it is applicable only to proteins containing free thiol groups located inside the membrane protein core, whereas only 66% of membrane proteins with known structure have thiol group(s), and only 91% of these cysteine residues are within the protein core. The presence of reducing agents commonly used in the protein purification process will interfere with thiol-CPM formation and reduce the signal-to-noise ratio. Reactivity of CPM is also pH dependent. 13 DSLS, on the other hand, is a label-free method and therefore not sensitive to buffer conditions. 4,14,15
In this study, we employed DSLS to measure the stability of membrane proteins and detect ligand binding. We used 3 different model proteins: CorA, a Mg++ channel protein from Thermotoga maritime; CpxA, a sensor protein (histidine kinase); and MsbA, an ABC (adenosine triphosphate [ATP] binding cassette) transporter from Escherichia coli. The CorA family of magnesium transporters is the primary Mg++ uptake system of most prokaryotes and a functional homologue of the eukaryotic mitochondrial magnesium transporter. Crystal structure of the full-length T. maritima CorA with bound Mg++ has been determined. 16 The transporter is a funnel-shaped homopentamer with 2 transmembrane helices per monomer. The channel is formed by an inner group of 5 helices and putatively gated by bulky hydrophobic residues. CpxA (histidine kinase, HK) is the inner membrane sensor of the Cpx pathway. Two-component systems that respond to particular stimuli by modifying the phosphorylation state of a cognate regulatory protein are the most prevalent form of signal transduction mediating the bacterial response to environmental signals, which is relayed by phosphorylation events. 17 MsbA is a bacterial ABC transporter. Its main role is the transport of lipid A, which is an essential component of the outer membrane of gram negatives. 18,19 Close homologues of MsbA have been identified in gram positives and mammals. Typically, MsbA homologues in gram positives confer resistance to drugs, whereas mammalian homologues of MsbA have been implicated in resistance of cancer cells to chemotherapeutic agents. The recent structures of MsbA show that this protein functions as a homodimer and most likely undergoes significant structural changes during its transport cycle. 20,21
Using these model proteins, our data confirm that DSLS can be used to assess thermostability of membrane proteins and to detect ligand binding, even in the presence of detergents.
Materials and Methods
Materials
Compounds listed in Table 1 were purchased from Sigma (St. Louis, MO), Fluka (St. Louis, MO), Calbiochem (La Jolla, CA), LC laboratories (Woburn, MA), and Sequoia Research P (Pangbourne, UK), as indicated along with the related catalogue numbers in the same table. The rest of the compounds used in this study are from Sigma unless stated otherwise.
The Library of Compounds
Expression and purification
E. coli CpxA and T. maritima CorA were subcloned in pET15b and expressed in BL21-Gold(DE3) from Stratagene (La Jolla, CA). 22 The membrane fractions were isolated from lysed cells, and proteins were solubilized in a solution containing 50 mM HEPES (pH 7.5), 500 mM NaCl, 5% glycerol, 10 mM imidazole, 1% n-dodecyl-β-d-maltoside (DDM; Anatrace, Maumee, OH), and protease inhibitors (Roche, Basel, Swizterland). Solubilized proteins were purified using Ni-NTA affinity resin (Qiagen, Valencia, CA). The hexahistidine tag was removed by adding 0.05 mg TEV protease (purified in SGC) per mg of purified protein. The TEV-protein mix sample was dialyzed overnight against dialysis buffer (50 mM HEPES [pH 7.5], 500 mM NaCl, 5% glycerol). The protein was then applied onto a second Ni-NTA column to isolate the pure untagged protein as described before. 22 The detergent concentration was reduced to 0.02% DDM by dialysis, and final buffer composition was 50 mM HEPES (pH 7.5), 500 mM NaCl, 5% glycerol, and 0.02% DDM, which was elevated upon protein concentration. 22
MsbA was subcloned in pET16 and expressed in E. coli BL21 (DE3) cells with a decahistidine tag using the autoinduction media. 23 Cells were harvested and membranes prepared by ultracentrifugation. Membranes were solubilized with 1% n-decyl-β-d-maltopyranoside (DM, Anatrace) in a buffer containing 50 mM Tris (pH 8.0), 500 mM NaCl, 10 mM imidazole, and 10% glycerol. Solubilized membranes were purified using Ni-NTA resin. Purified MsbA was exchanged into 20 mM Tris (pH 8), 500 mM NaCl, and 0.2% DM by size exclusion chromatography.
Thermal stability studies by DSLS
The thermal stability of membrane proteins was studied by differential static light scattering using StarGazer (Harbinger Biotechnology and Engineering Corporation, Markham, Canada). This method assesses protein stability by monitoring protein aggregation during protein denaturation. MsbA at 0.5 mg/mL and CorA and CpxA at 1 mg/mL in a 50-µL volume in a clear-bottom 384-well plate (Nunc) were heated from 25 to 85 °C at 1 °C/min. Wells were covered with mineral oil to prevent evaporation. Protein aggregation was monitored by tracking the change in scattered light that was detected by a CCD camera. 8 Images of the plate were taken every 0.5 °C. The pixel intensities in a preselected region of each well were integrated using image analysis software to generate a value representative of the total amount of scattered light in that region. These intensities were then plotted against temperature for each sample well and fitted to obtain the aggregation temperature (Tagg) as described before. 8
Small library of compounds used to screen MsbA
A library of 39 physiological and drug-like compounds was selected and prepared at final concentrations of 5 and 0.1 mM, respectively, to screen MsbA. The final DMSO (Sigma) concentration was 1%.
Results and Discussion
A high percentage of soluble proteins aggregate following heat denaturation due to the exposure of buried hydrophobic groups (reviewed by Bischof and He, 24 Dobson, 25 and Murphy and Kendrick 26 ), and the aggregates can be detected by DSLS. 4,27-29 Using DSLS to detect thermodenaturation of isolated membrane proteins reconstituted in detergent micelles requires that the denaturing proteins aggregate subsequent to the heat-induced unfolding. At difference with soluble proteins, the aggregation of membrane proteins could be affected and interfered by the presence of detergent micelles contained in the protein preparations. Heating low detergent concentrations above the critical micellar concentration (CMC) usually causes marked changes in basic properties such as viscosity and light and neutron scattering, which are indicative of a change in micellar size and/or shape. 30,31 If these changes are detected by DSLS, they may have a significant interference during the protein denaturation detection. We applied a DSLS to test 5 different detergents commonly used during membrane protein preparations (DM, DDM, n-dodecylphosphocholine [FC12], lauryldimethylamine-N-oxide [LDAO], and ANZERGENT®) and 3 membrane proteins (MsbA, CpxA, and CorA). We observed that DSLS did not detect physical changes occurring during the scanning of the detergents alone but detected the aggregation of the 3 tested proteins, which showed sigmoidal aggregation curves similar to the curves usually observed during scanning of soluble proteins ( Fig. 1 ). 4,8,32 The detergents alone, at 4 times CMC, did not increase the DSLS background and did not change the light-scattering signal when the temperature was increased. The Tagg value of the protein depends on type and concentration of the detergent. Screening MsbA in different concentrations of DM indicates that the presence of the detergent stabilizes the protein. However, the Tagg values are similar as long as the detergent concentration is above the CMC value ( Fig. 2 ). In general, the effect of detergent concentration on protein stability may vary for different detergents or proteins, and different preparations of a membrane protein may show some variation in absolute Tagg values. However, considering the relative stability of a protein from a common batch against different buffers or added ligands is more useful. All together, these observations indicate that DSLS can be used to study the stability of membrane proteins with no interference from the presence of detergents.
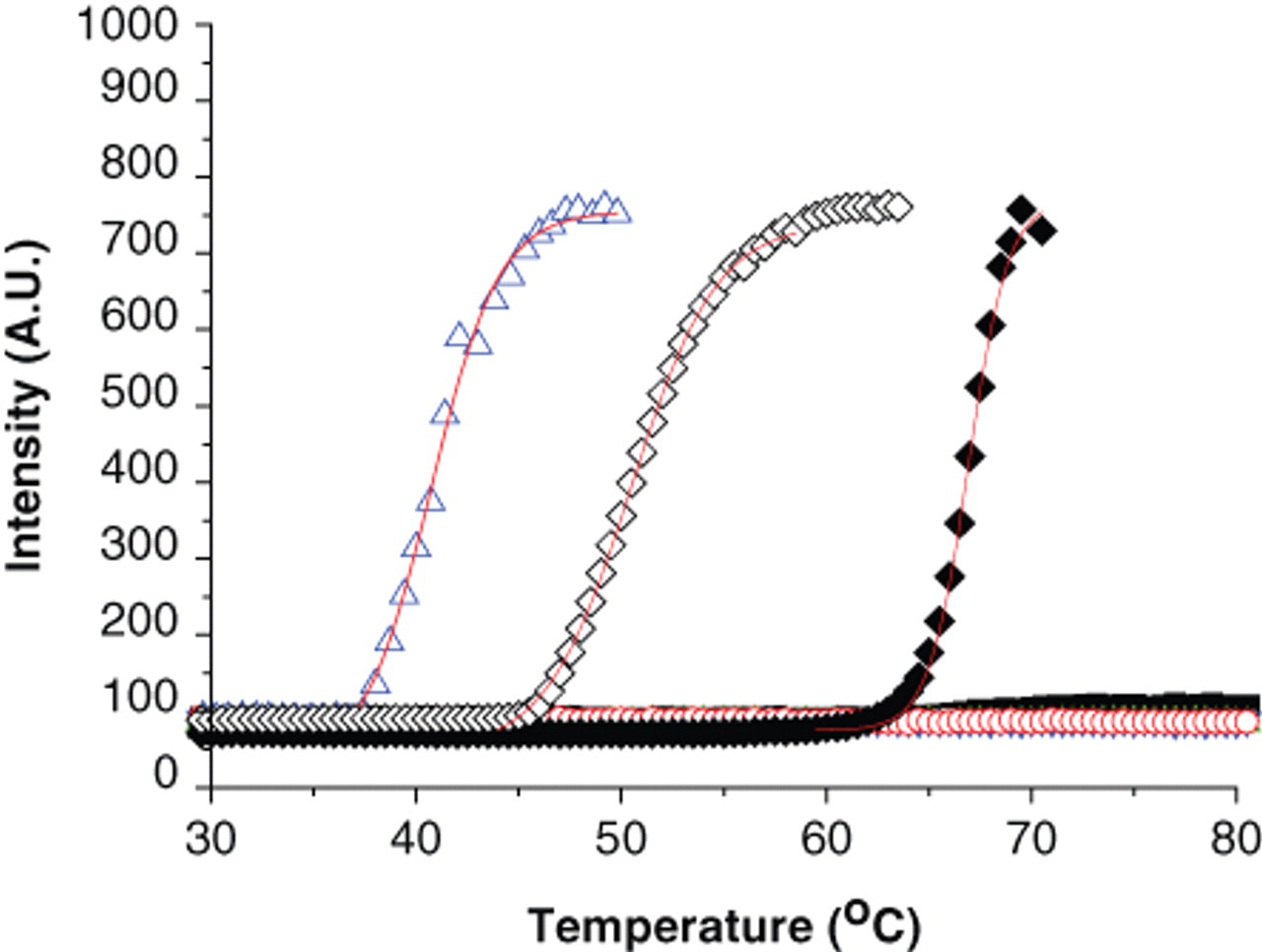
Thermodenaturation of membrane proteins in the presence of detergents. CpxA (◊) and CorA (♦) at 1 mg/mL in n-dodecyl-β-d-maltoside (DDM; 2× critical micellar concentration [CMC]) and MsbA (Δ) at 0.5 mg/mL in DM (2× CMC) were screened by differential static light scattering (DSLS). The solid line represents the best fit for each curve using the Boltzmann equation. n-decyl-β-d-maltopyranoside (DM; ■), DDM (●), n-dodecylphosphocholine (FC12; ▲), ANZERGENT® (□), and lauryldimethylamine-N-oxide (LDAO; ○) at 4× CMC did not change the background and were not detected by DSLS.
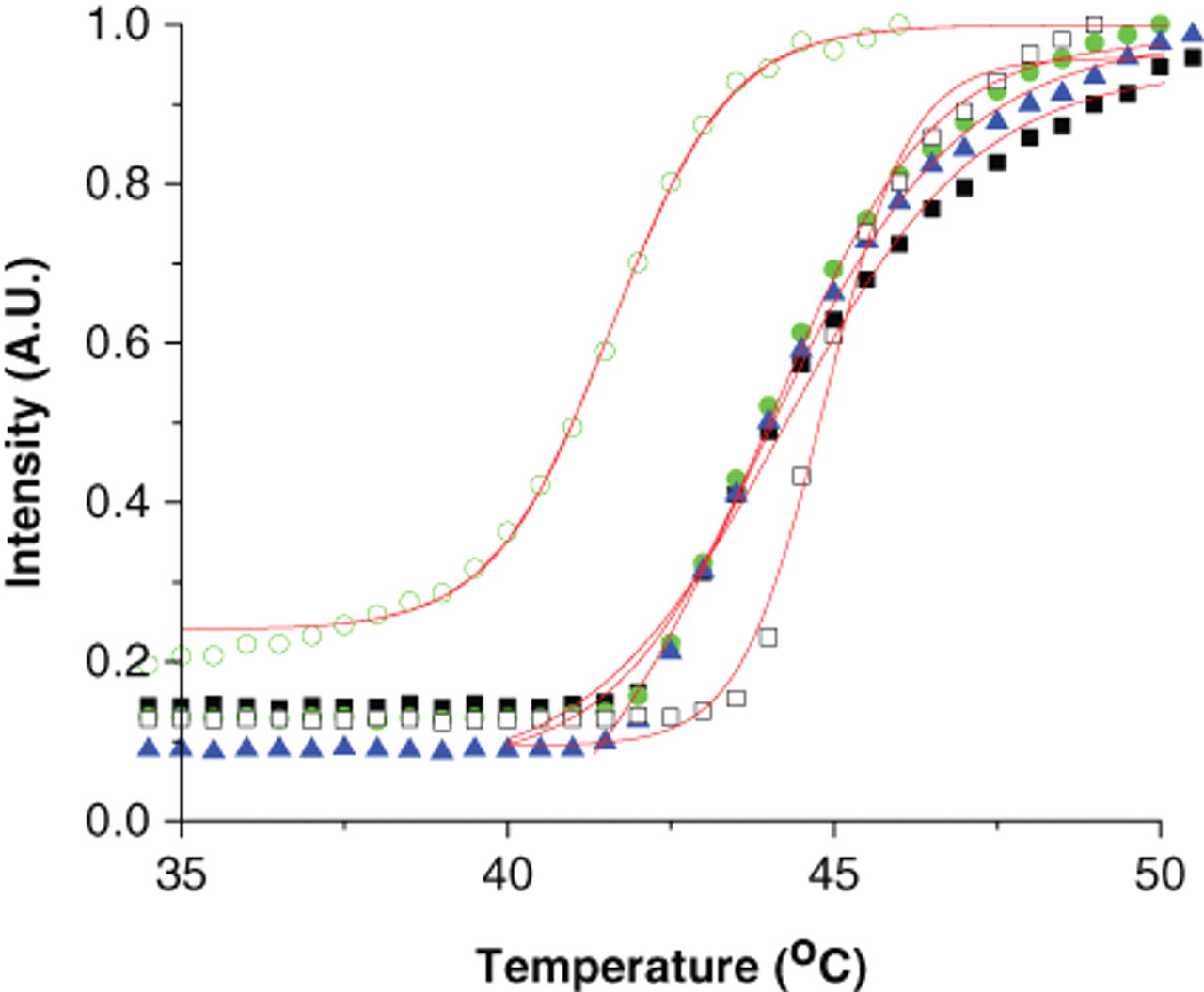
Effect of concentration of n-decyl-β-d-maltopyranoside (DM) on stability of MsbA. Differential static light scattering (DSLS) was performed on MsbA in DM at 0.25 (○), 0.5 (□), 1 (▲), 2 (●), and 4 (■) times critical micellar concentration.
Detection of ligand binding
To demonstrate the feasibility of DSLS to study the stability of membrane proteins, we investigated whether this method has the potential to detect stabilization induced by the binding of small molecules in the same way as it is currently used for soluble proteins. For this purpose, the 3 selected membrane proteins—MsbA, CpxA, and CorA—were screened in the presence of some of their known ligands and a few other compounds as negative controls ( Fig. 3 ). MsbA with a transition temperature (Tagg) of 40 °C was stabilized by 4.5 °C in the presence of 2 mM ATP ( Fig. 3A ), and CpxA was stabilized by 5 °C in the presence of 10 mM ATP but was not affected by nicotinamide adenine dinucleotide (NADH), which was not expected to bind ( Fig. 3B ). Interestingly, CorA was stabilized by the divalent cations, Mg++ and Ca++, as expected but not Li+, indicating that DSLS can detect specific ligand binding ( Fig. 3C ). To further evaluate the sensitivity of DSLS in detecting ligands, different ligands were titrated against these proteins. Lipid A, a known substrate for MsbA, stabilized this protein in a concentration-dependent manner with as much as 4 °C at a 100-µM concentration ( Fig. 4A ). Increase in the stability of wild-type MsbA upon increase in concentration of ATP was also detectable ( Fig. 4B ). A stabilizing effect of ATP on CpxA was also concentration dependent ( Fig. 4C ). CorA is known to bind and transport Mg++, and its structure with an Mg++ ion bound between monomers at a conserved site in the cytoplasmic domain has been determined. 16 The structure of CorA in complex with Ca++ has also been reported. 33 DSLS was also able to detect the concentration- dependent stabilizing effect of Mg++ and Ca++ on this protein ( Fig. 4D ). However, increasing concentration of Li+ up to 5 mM did not affect the DSLS signal, indicating that CorA does not bind to Li+ (data not shown). Our data clearly indicate that DSLS is a method that can detect the stabilizing effect of ligands upon binding to membrane proteins.

Stabilization effect of ligands on MsbA, CpxA, and CorA. Stability of (); and ( ), and 1 mM Mg++ (▲) and 1 mM Li+ (∇) were assessed by differential static light scattering.
), and 1 mM Mg++ (▲) and 1 mM Li+ (∇) were assessed by differential static light scattering.
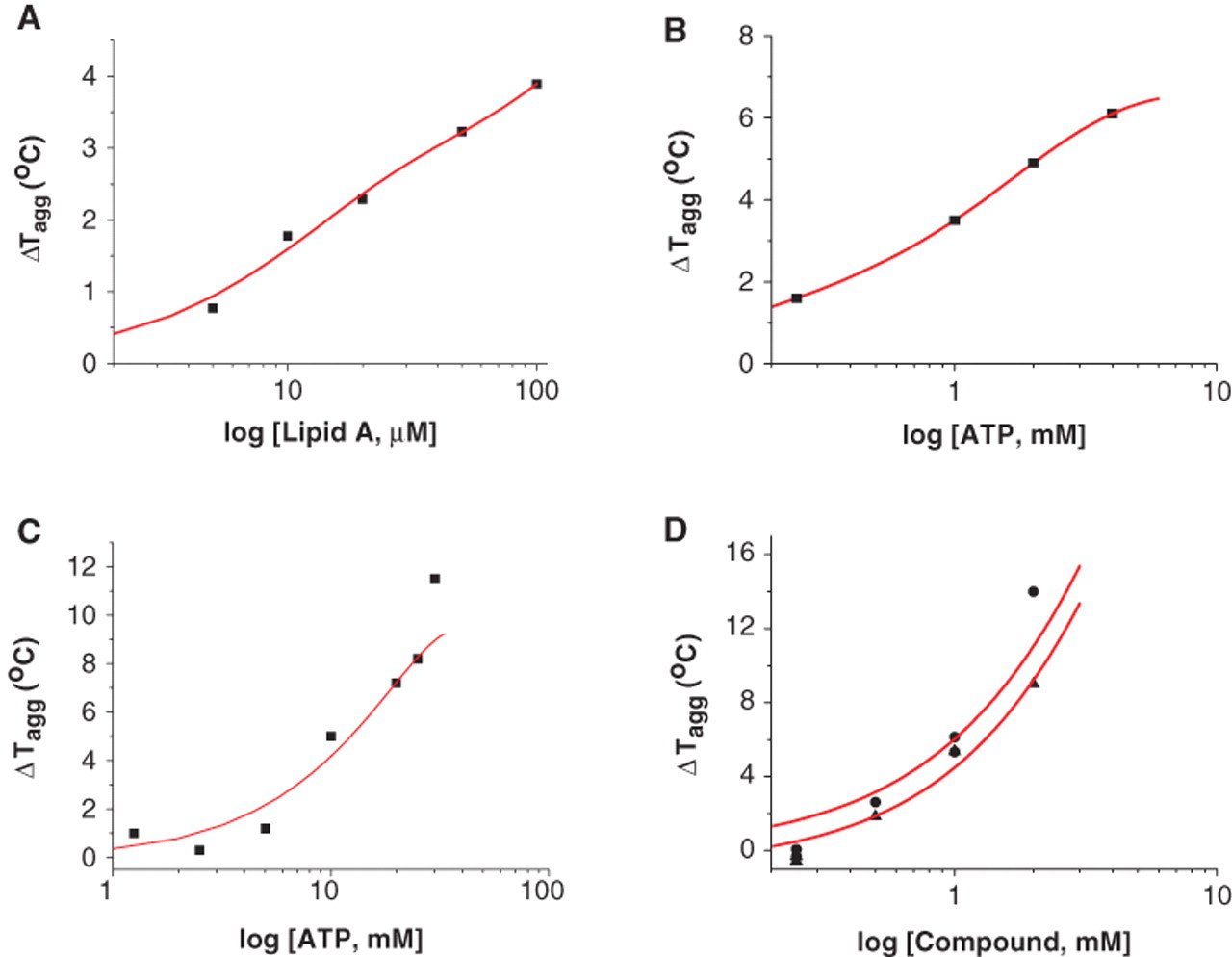
Effect of concentration of ligands on stability of MsbA, CpxA, and CorA. Effect of increasing concentrations of (
Screening for ligands in the 384-well format
The ability to screen membrane proteins against a larger number of compounds will facilitate finding ligands and may result in a higher rate of success in preparing membrane proteins and determining their crystal structure. In this study, we investigated the possibility of employing DSLS to screen membrane proteins against libraries of compounds in a 384-well format. We designed a small library of 39 compounds, including drugs and physiological molecules ( Table 1 ), and screened wild-type ( Fig. 5A ) and an ATPase-deficient mutant of MsbA (E506Q; Fig. 5B ) against this library. Five compounds significantly stabilized both the wild-type and mutant proteins: ADP, ATP, acetyl-CoA, β-γ-methyladenosine-5′-triphosphate, and AMP-PMP. However, NADP+ and NADPH stabilized the wild-type MsbA but not the mutant by more than 2 °C. Our results showed that the MsbA mutant was capable of binding ATP, consistent with previous studies. 34 To examine the reproducibility of detecting changes in the stability of membrane proteins and detect ligands, we screened multiple samples (36) of wild-type MsbA in the presence and absence of 5 mM ATP (total of 72 samples). We were able to detect binding of ATP reproducibly with a Z′ factor of 0.75 ( Fig. 6 ). Although the number of samples in this experiment was low and did not cover a full 384-well plate, our data prove that DSLS can be used to screen membrane proteins for ligands in a 384-well format.
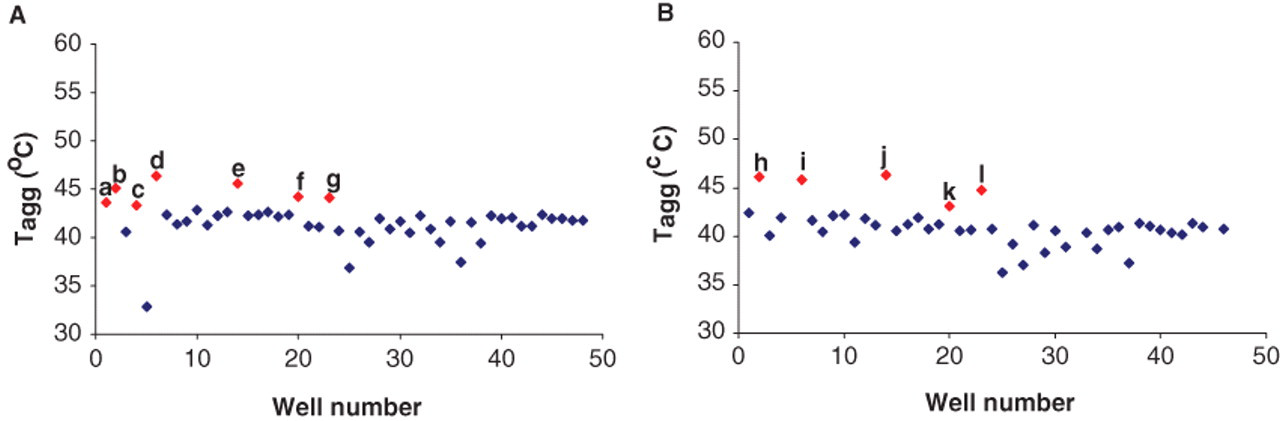
Screening the wild-type MsbA and MsbA E506Q mutant against a library of 39 compounds. (
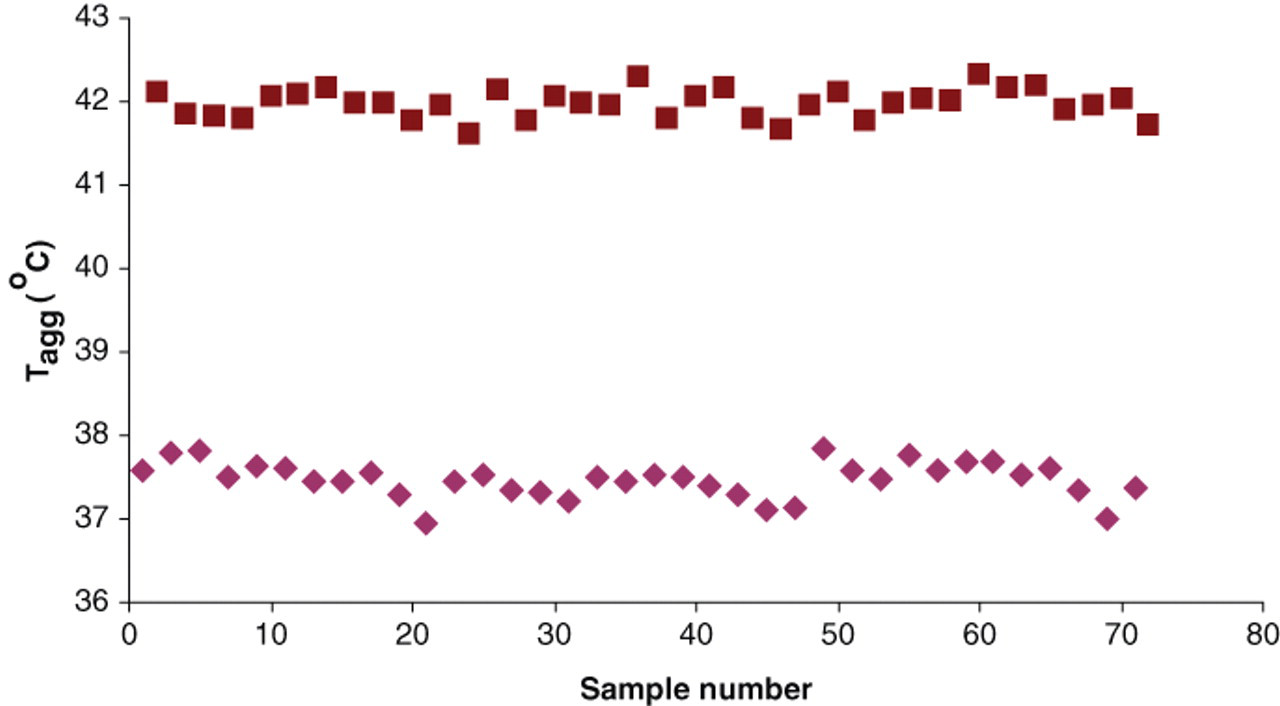
Reproducibility of hit detection in a 384-well format. Stability of wild-type MsbA in the presence (■) and absence (♦) of 5 mM adenosine triphosphate was assessed within a 384-well plate. Samples were distributed within the plate 36 times each. A Z′ factor of 0.75 was calculated as described by Zhang et al. 35
Concluding remarks
Our results indicate that DSLS can be used to assess the stability of membrane proteins, screen them for optimum buffer and detergents, and detect the stabilization effect of ligands upon binding selectively. Such screening can be performed reproducibly in a 384-well format, which is crucial in facilitating identification of ligands and functional and structural studies of membrane proteins.
Footnotes
Acknowledgements
The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust.
GGP was supported by a grant from the Canadian Institutes for Health Research. HG was supported by a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship.