
Abstract
mGluR1 antagonists have been postulated to be novel CNS drugs, including antipsychotics. Toward this end, the authors developed a β-lactamase reporter assay to identify mGluR1 antagonists. β-Lactamase has several interesting features for high-throughput screening, including very high sensitivity and less well-to-well variation than other reporter enzymes. mGluR1-expressing Chinese hamster ovary (CHO) cells with the β-lactamase gene under control of the nuclear factor of activated T cells (NFAT) promoter (CHO-NFAT-bla-hmGluR1b) exhibited very high basal activity, resulting in an inadequate signal-to-basal (S/B) ratio. Coexpression of glutamate/aspartate transporter (GLAST) with mGluR1 in the cell line (CHO-NFAT-bla-hmGluR1b-GLAST) dramatically decreased basal activity and improved the S/B ratio (from 2- to 20-fold). The contribution of GLAST to lowering basal activity and increasing the S/B ratio was validated by the expression level of GLAST mRNA and by a GLAST inhibitor. Antagonistic activities of known mGluR1 antagonists in the β-lactamase reporter assay were comparable with those in the conventional Ca2+ mobilization assay. The Z′ factor of the β-lactamase reporter assay was 0.89 under optimized conditions. Taken together, the β-lactamase reporter assay with CHO-NFAT-bla-hmGluR1b-GLAST could be a novel high-throughput assay for mGluR1 antagonist screening. This is the first description of a successful β-lactamase reporter assay among all mGluR subtypes.
Keywords
Introduction
M
Earlier efforts to identify an mGluR1-selective antagonist by chemical library screening using an orthosteric radioligand binding assay were unsuccessful. This was presumably due to the highly conserved amino acid sequences of the L-glutamate-binding site in mGluR1 and mGluR5. 2 On the other hand, a functional assay based on the detection of intracellular Ca2+ increases mediated via receptor activation has allowed us to identify mGluR1-selective allosteric antagonists. 3-5 The success of this approach may stem from the fact that functional assays can identify compounds interacting with sites different from the L-glutamate binding site, such as the transmembrane domain. These sites are less conserved than the L-glutamate binding site. Several animal studies using these mGluR1-selective allosteric antagonists indicate that blockage of mGluR1 could ameliorate CNS disorders, including neuropathic pain, neurodegeneration, and psychiatric diseases. 6-8 However, it is not known whether mGluR1 is involved in human CNS disorders.
In addition to the Ca2+ mobilization assay, a reporter gene assay under the control of the NFAT promoter (which is activated by an increase in intracellular Ca2+) is another functional assay applicable to the identification of Gq-protein-coupled mGluR1 ligands. Of the known reporter gene assays, β-lactamase has several interesting features for high-throughput screening (HTS). 9 For example, this reporter assay uses β-lactamase and its cell-permeable fluorogenic substrate, CCF2. Excitation of CCF2 produces green fluorescence mediated via fluorescence resonance energy transfer (FRET). Cleavage of CCF2 by β-lactamase causes loss of FRET and results in blue fluorescence upon excitation. This wavelength shift is detectable in individual cells containing only 100 β-lactamase molecules, suggesting that the assay has very high sensitivity. 10 Furthermore, ratiometric data analysis of the CCF2 substrate (the ratio of the net blue fluorescence signal intensity and the green fluorescence signal intensity) eliminates problems arising from well-to-well variations in cell number and fluorescence signal intensity. High sensitivity and decreased variation enable this assay to be used in an ultra-high-throughput assay format. 11-13 Therefore, the β-lactamase reporter gene under the control of the NFAT promoter has been applied to ligand screening for many Gq-protein-coupled receptors such as oxytocin receptor, 13 bradykinin B1 receptor, 14 gonadotropin-releasing hormone receptor, 15 and M1 muscarinic receptor. 16 However, there is no report of a β-lactamase reporter assay for family C G-protein-coupled receptors such as mGluR1, which possess an unusually large N-terminal extracellular domain and allosteric binding sites in the transmembrane domain.
In the present study, we developed a β-lactamase reporter assay for mGluR1. Stable expression of mGluR1 in Chinese hamster ovary (CHO) cells with the β-lactamase gene under the control of the NFAT promoter exhibited very high basal activity, making this approach unsatisfactory for HTS. However, coexpression of glutamate/aspartate transporter (GLAST) with mGluR1 in the cell line dramatically decreased basal activity and improved the signal-to-basal (S/B) ratio. Antagonist potencies of mGluR1 antagonists in the β-lactamase reporter assay were compared with those in the Ca2+ mobilization assay. This is the first description of a successful β-lactamase reporter assay for mGluR. This assay is a promising alternative high-throughput assay, with high sensitivity and less variation than existing assays for mGluR1 antagonist screening.
Materials and Methods
Materials
L-glutamate was purchased from Sigma-Aldrich (St. Louis, MO). Trans-PDC (L-trans-pyrrolidine-2,4-dicarboxylic acid) and CPCCOEt (7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester) were purchased from Tocris Bioscience (Bristol, UK). LY456066 (2-[4-(Indan-2-ylamino)-5,6,7,8-tetrahydro-quinazolin-2-ylsulfanyl]-ethanol, HCl), R214127 (1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenyl-1-ethanone), YM-298198 (6-amino-N-cyclohexyl-N,3-dimethylthiazolo[3,2-a]benzimidazole-2-carboxamide), compound 1 (2RS-2-cyclopentyl-7-fluoro-5-{[3-(4-methylpiperazin-1-yl)propyl]amino}indan-1-one), and compound 2 (tert-butyl 4-[1-(2-fluorophenyl)-1H-1,2,3-triazol-4-yl]-3,6-dihydropyridine-1(2H)-carboxylate) were synthesized in-house (see supplemental materials at http://jbx.sagepub.com/supplemental). L-proline was purchased from Wako Pure Chemical Industries (Osaka, Japan). Dialyzed fetal bovine serum, culture media, and other reagents used for cell culture were purchased from Invitrogen (Carlsbad, CA). All other reagents used were of molecular or analytical grade where appropriate.
Methods
Cells
Human mGluR1b cDNA was obtained as described previously. 4 CHO-NFAT-bla cells were licensed and obtained from Aurora Biosciences (San Diego, CA). CHO-NFAT-bla cells were transfected with human mGluR1b cDNA cloned into pcDNA3.1hyg (Invitrogen) and selected in Dulbecco’s modified Eagle’s medium (DMEM) with 10% dialyzed fetal bovine serum, 100 units/mL penicillin, 100 units/mL streptomycin, 1% proline, and 250 µg/mL zeocin (Invitrogen), supplemented with 250 µg/mL hygromycin B (Invitrogen) at 37°C with 5% CO2 in a humidified atmosphere. The stable cell lines were isolated and selected by their ability to elicit Ca2+ mobilization following L-glutamate addition.
Rat GLAST cDNA was obtained from rat brain poly A+ RNA (Clontech, Palo Alto, CA) using RT-PCR and cloned into pcDNA3. This GLAST cDNA encoded a peptide sequence identical to the GLAST sequence reported previously. 17 The CHO-NFAT-bla cell lines stably expressing human mGluR1b were transfected with GLAST cDNA cloned into pIRESneo (Clontech) and selected in the above medium supplemented with 500 µg/mL geneticin (Invitrogen). The stable cell lines were isolated and selected by their ability to induce β-lactamase activity following L-glutamate addition.
Ca2+ mobilization assay
Intracellular Ca2+ mobilization was measured as described previously. 4 Briefly, cells were seeded at 5 × 104 cells/well in a 96-well black-well/clear-bottom plate (PerkinElmer Life and Analytical Sciences, Boston, MA) and cultured overnight. The cells were then incubated with 4 µM Fluo-3 in assay buffer (Hanks’s balanced salt solution containing 20 mM HEPES and 2.5 mM probenecid) containing 1% dialyzed fetal bovine serum for 1 h at 37°C with 5% CO2 in a humidified atmosphere. The extracellular dye was removed, and Ca2+ flux was measured using a fluorometric imaging plate reader (FLIPR; Molecular Devices, Sunnyvale, CA). Compounds were preincubated for 5 min before addition of L-glutamate. After preincubation, the antagonistic activities of compounds were evaluated for 3 min after addition of an agonist. The final concentration of L-glutamate was 30 µM in the antagonist assay. IC50 values of compound 1 toward human mGluR1a and mGluR5 were obtained as reported previously. 4
β-lactamase assay
On the day of the assay, the cells were rinsed with phosphate-buffered saline (PBS) and incubated with enzyme-free cell dissociation buffer (Invitrogen) for about 15 min at 37°C with 5% CO2 in a humidified atmosphere. Detached cells were rinsed and suspended at 1 × 106 cells/mL with assay medium (phenol red-free DMEM supplemented with 25 mM HEPES). Antagonists (17 µL of 6 × final concentration) or DMSO were added to the wells of a 96-well black-well/clear-bottom plate (Coaster, Corning, NY), and then 50 µL of cell suspension (50,000 cells/well) was plated into the 96-well plate, followed by 5-min incubation with the antagonists. For agonist stimulation, 33 µL of 3 × final concentration of L-glutamate (final 50 µM) was added to the cells in the 96-well plate, followed by incubation for 5 h at 37°C with 5% CO2 in a humidified atmosphere, unless stated otherwise. The 6 × final concentration of CCF2/AM substrate loading buffer was prepared according to the loading protocol of Aurora Biosciences by combining 12 µL of solution A (1 mM CCF2/AM in DMSO), 60 µL of solution B (100 mg/mL pluronic F-127 in DMSO containing 0.1% acetic acid), 925 µL of solution C (24% polyethylene glycol-400 with 12% enhanced substrate solution, Aurora Biosciences or Invitrogen), and 75 µL of solution D (15 mM probenecid in 200 mM NaOH). To determine β-lactamase activity, 20 µL of 6 × final concentration of CCF2/AM substrate loading buffer was added to the cells, and then the cells were incubated in the dark at room temperature for 2 h, unless stated otherwise. The plate was read in a fluorescence plate reader (CytoFluor 4000, PerSeptive Biosystems, MA) using 409-nm excitation and 460-nm (blue) and 530-nm (green) emissions through the clear bottom of the plate. The data were plotted as a “blue/green ratio” (460 nm/530 nm) after background subtraction (medium only, without cells).
Determination of gene expression level using real-time PCR
Total RNA was extracted from the cells using an RNeasy Mini kit (Qiagen, Hilden, Germany). Real-time PCR was carried out using a modification of a previously described method. 18 TaqMan Gene Expression Assays (Assay ID; Rn00570130_m1) and TaqMan Rodent GAPDH Control Reagents (Applied Biosystems, Foster City, CA) were used to quantify mRNA expression of GLAST and GAPDH, respectively. Reverse transcription of total RNA (1 µg) was performed in a total volume of 20 µL using random hexamers and a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s protocol. The resultant cDNA sample (20 µL) was diluted with 80 µL of nuclease-free water. Quantitative real-time PCR was performed in a total volume of 10 µL containing 5 µL of qTaq DNA polymerase mix (Clontech), 0.5 µL of TaqMan probe/primers mixture, and 0.6 µL of the diluted cDNA templates, using an ABI Prism 7900 cycler (Applied Biosystems). The cycling profile was 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C, as recommended by the manufacturer.
Data analyses and statistics
Data analyses were performed using Prism (Version 4.03) from GraphPad Software (San Diego, CA). Concentration-response curves for Ca2+ mobilization and β-lactamase activity were fitted using nonlinear regression analysis. Student t-test was used to analyze the effectiveness of removal of L-glutamine in the β-lactamase assay. A probability level of <0.05 was considered statistically significant. The Z′ factor, the index for assay quality assessment in assay development and optimization, was determined using the following equation 19 : Z′ = 1 − (3σC+ + 3σC−)/│µC+ − µC−│, where µC+ and µC− are the means of positive control signal and negative control signal, respectively, and σC+ and σC− are the standard deviations of the corresponding signals.
Results
Optimization of a β-lactamase assay using CHO-NFAT-bla cells expressing human mGluR1
CHO-NFAT-bla cells induce β-lactamase activity via activation of the NFAT promoter following increases in intracellular Ca2+ elicited by the activation of mGluR1. Induced β-lactamase cleaves CCF2, resulting in increased blue fluorescence ( Fig. 1 ). In order to set up a β-lactamase assay for mGluR1, CHO-NFAT-bla was transfected with mGluR1b cDNA cloned into pcDNA3.1hyg, and the cell line stably expressing mGluR1 (CHO-NFAT-bla-hmGluR1b) was cloned. To confirm the expression of functional mGluR1, Ca2+ response elicited by agonist stimulation was evaluated in CHO-NFAT-bla-hmGluR1b. The cell line exhibited a robust increase in intracellular Ca2+ concentration following the addition of 1 mM L-glutamate ( Fig. 2A ). In the β-lactamase assay, a higher basal level of β-lactamase activity (1.8 ± 0.44, n = 3) was observed in the cell line compared with the host cells (0.27 ± 0.056, n = 4). Furthermore, 1 mM L-glutamate did not significantly induce β-lactamase activity. During the course of optimizing the β-lactamase assay, removal of L-glutamine from the assay medium was found to contribute to a lower basal activity (0.88 ± 0.19, n = 3) and an increase in β-lactamase activity (1.8 ± 0.23, n = 3) following L-glutamate stimulation, whereas the S/B ratio remained low (2.1 ± 0.27, n = 3; Fig. 2B ).
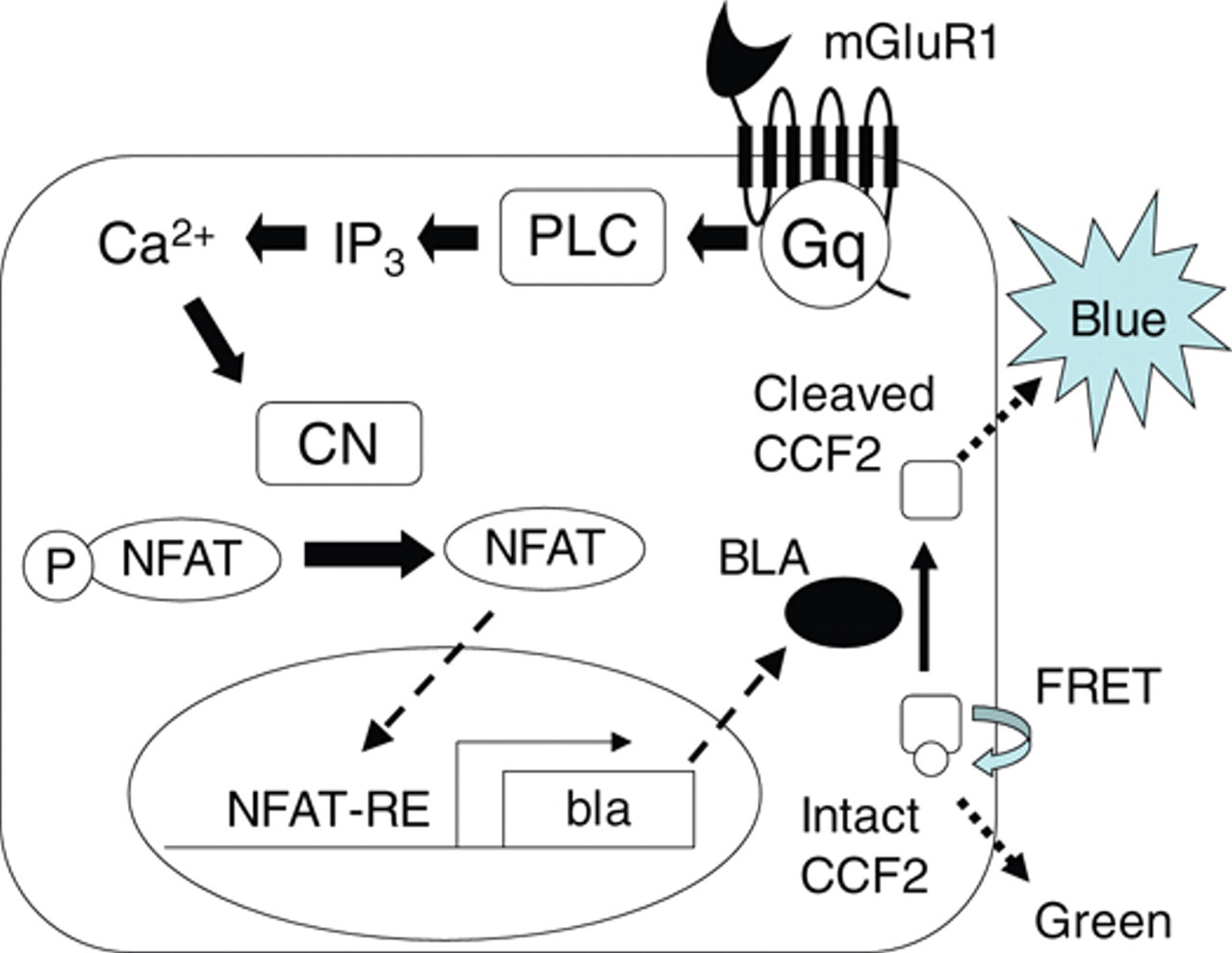
A schematic diagram of the β-lactamase assay for mGluR1. Activation of mGluR1 stimulates phospholipase C (PLC) via Gq-protein. Activated PLC produces inositol-1,4,5-trisphosphate (IP3) and elicits subsequent increases in intracellular Ca2+. Calcineurin (CN) activated by Ca2+ dephosphorylates cytoplasmic phosphorylated NFAT (P-NFAT), allowing it to move into the nucleus. NFAT translocated into the nucleus activates NFAT–response element (NFAT-RE) to induce β-lactamase gene (bla) expression. Intact CCF2 produces green fluorescence via fluorescence resonance energy transfer (FRET). CCF2 cleaved by β-lactamase protein (BLA) loses FRET and results in blue fluorescence.
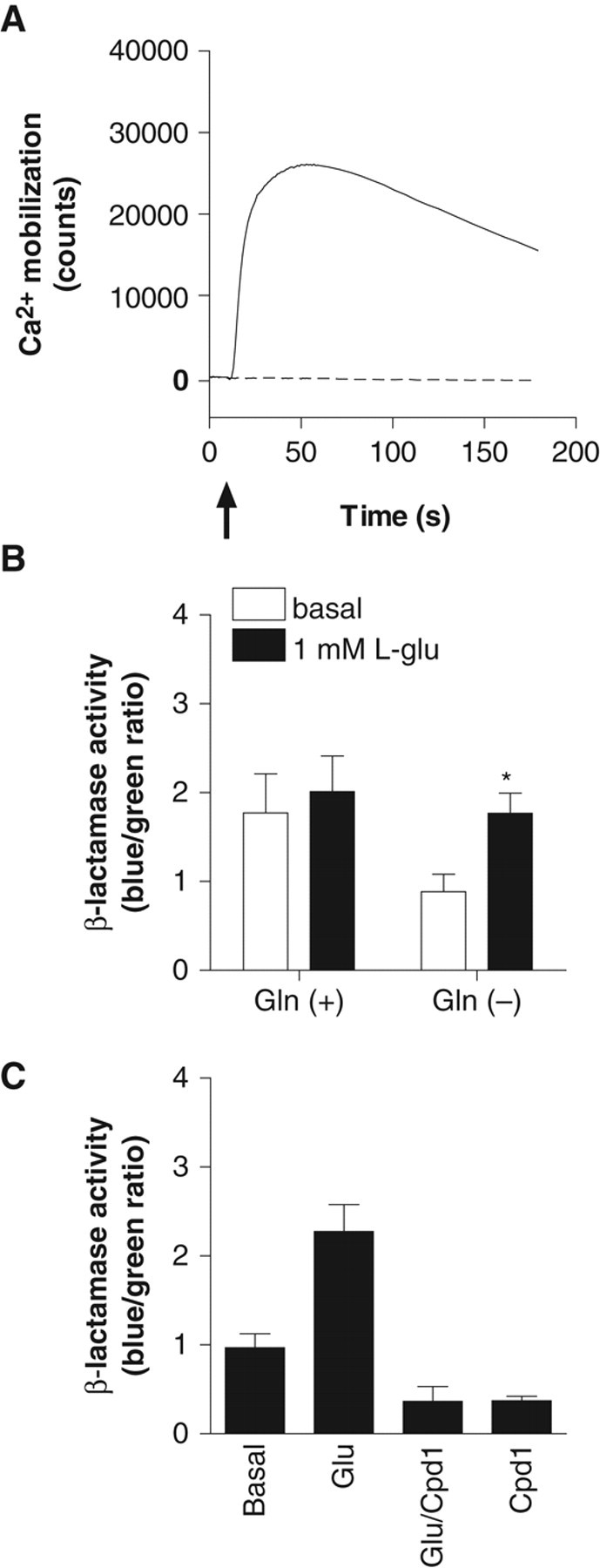
Pharmacological characterization of CHO-NFAT-bla-hmGluR1b. (
Compound 1 is an mGluR1-selective antagonist with IC50 values (nM) of 36 ± 4.5 and >10,000 (n = 6) against L-glutamate-induced Ca2+ mobilization in CHO cells expressing human mGluR1a and CHO cells expressing human mGluR5, respectively, as assayed by FLIPR. Compound 1 at 10 µM inhibited β-lactamase activity induced by 1 mM L-glutamate to below basal levels. In addition, basal β-lactamase activity was also inhibited by compound 1 in the absence of L-glutamate stimulation ( Fig. 2C ).
The decreased basal β-lactamase activity caused by the mGluR1 antagonist suggested that mGluR1 was still activated in the absence of exogenously added L-glutamine and L-glutamate. Endogenous L-glutamate is known to be released from CHO cells and to activate mGluR. 20 We therefore investigated the effect of removing L-glutamate on the β-lactamase assay. To remove L-glutamate from the assay, CHO-NFAT-bla-hmGluR1b cells were transfected with glutamate transporter, GLAST cDNA cloned into a mammalian expression vector. Expression of GLAST decreased the basal level of β-lactamase activity (0.37 ± 0.010, n = 3) compared with mock cells transfected with the control vector (1.0 ± 0.032, n = 3). The lower basal β-lactamase activity in cells coexpressing mGluR1b with GLAST was similar to that in the presence of 10 µM compound 1 (0.37 ± 0.0052, n = 3; Fig. 3 ). A series of optimization experiments for the β-lactamase assay revealed that removal of L-glutamine from the assay medium and uptake of L-glutamate by coexpression of GLAST helped reduce the basal level and improve the SB ratio.
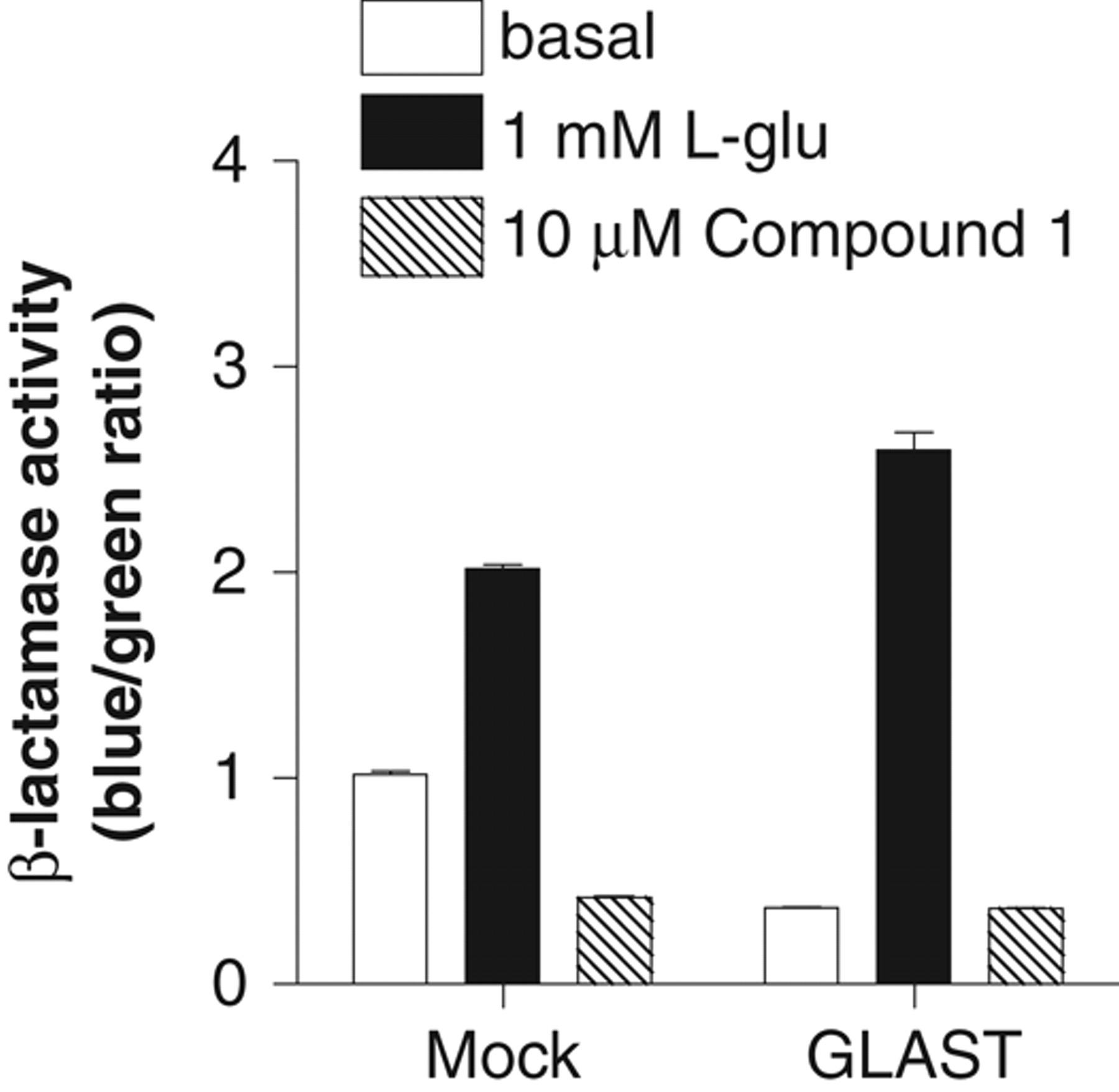
Effect of glutamate/aspartate transporter (GLAST) expression on β-lactamase activity in CHO-NFAT-bla-hmGluR1b. CHO-NFAT-bla-hmGluR1b cells were transfected with GLAST cDNA cloned into pIRESneo (GLAST) or pIRESneo control vector (Mock). The cells were cultured in medium with 500 µg/mL geneticin for 2 to 3 weeks and then were used for the β-lactamase assay. Open and closed bars represent β-lactamase activity in the absence or presence of 1 mM L-glutamate. Hatched bars indicate β-lactamase activity in the presence of 10 µM of the mGluR1 antagonist, compound 1. All data are the means ± SEM from 3 individual experiments performed in duplicate or triplicate.
Pharmacological characterization of CHO-NFAT-bla cells coexpressing human mGluR1 with GLAST
To obtain CHO-NFAT-bla cells stably coexpressing mGluR1 with GLAST, CHO-NFAT-bla-hmGluR1b was transfected with GLAST cDNA cloned into pIRESneo, and the G418-resitant clones were evaluated using the β-lactamase assay. One representative clone, referred to as CHO-NFAT-bla-hmGluR1b-GLAST, was pharmacologically characterized. The basal level of β-lactamase activity (0.14 ± 0.012, n = 3) in CHO-NFAT-bla-hmGluR1b-GLAST was much lower than that of the parent clone, CHO-NFAT-bla-hmGluR1b (0.76 ± 0.14, n = 4). In CHO-NFAT-bla-hmGluR1b-GLAST, L-glutamate increased β-lactamase activity in a dose-dependent manner, with an EC50 value of 30 ± 1.7 µM (n = 3; Fig. 4A ). The EC50 value for CHO-NFAT-bla-hmGluR1b was 17 ± 3.3 µM (n = 4). The S/B ratio was dramatically improved, from 2.3 ± 0.2 to 20 ± 1.5, by coexpression of GLAST in the cell line. The Hill coefficients for L-glutamate in the β-lactamase assay were 1.2 ± 0.14 (n = 4) and 2.8 ± 0.18 (n = 3) in CHO-NFAT-bla-hmGluR1b and CHO-NFAT-bla-hmGluR1b-GLAST, respectively. In the Ca2+ mobilization assay, CHO-NFAT-bla-hmGluR1b and CHO-NFAT-bla-hmGluR1b-GLAST elicited comparable increases in intracellular Ca2+ concentration ( Fig. 4B ). The EC50 values of L-glutamate in CHO-NFAT-bla-hmGluR1b and CHO-NFAT-bla-hmGluR1b-GLAST were 5.7 ± 1.7 and 8.0 ± 0.69 µM (n = 3), respectively. The Hill coefficients for L-glutamate in the Ca2+ mobilization assay were 1.5 ± 0.31 (n = 3) and 2.7 ± 0.35 (n = 3) in the absence and presence of GLAST, respectively.
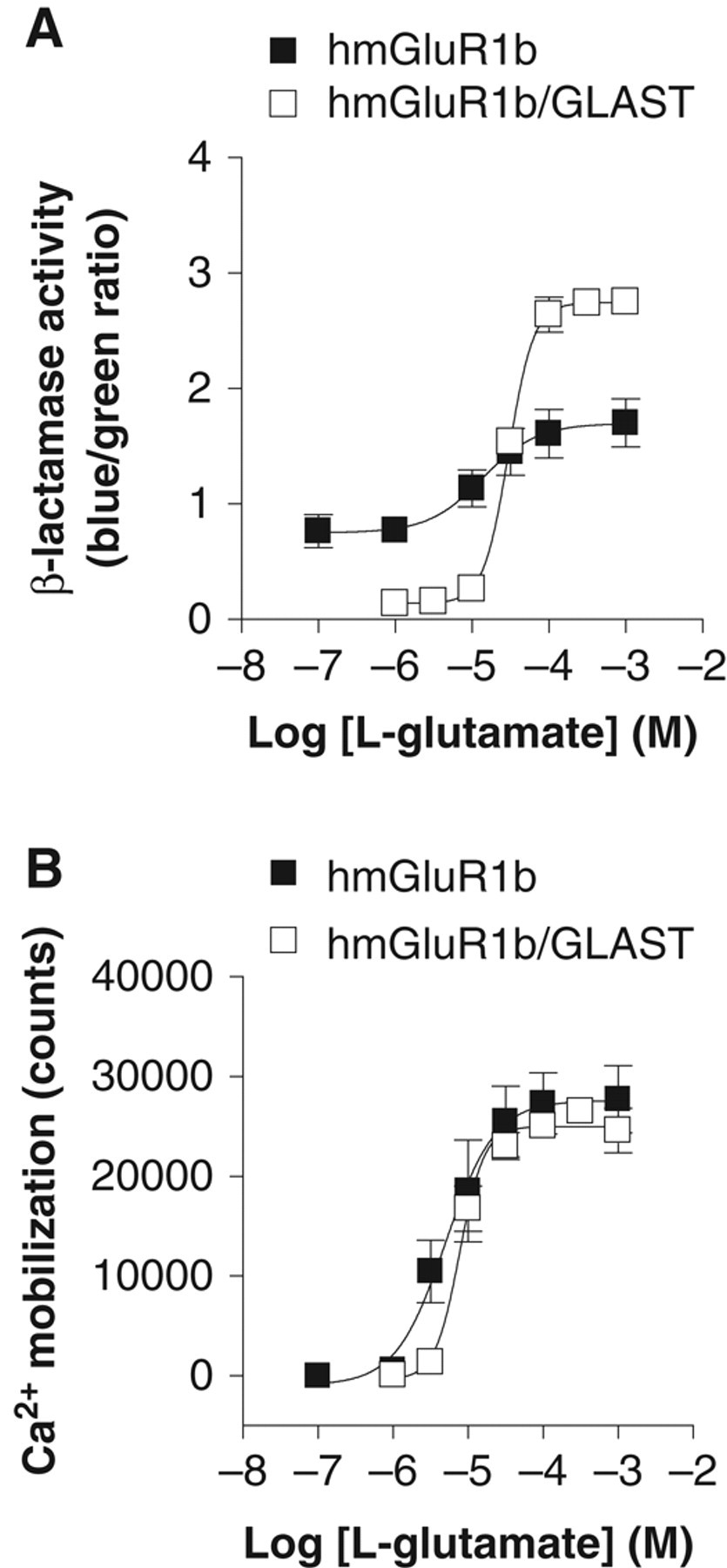
Comparison between the CHO-NFAT-bla-hmGluR1b and CHO-NFAT-bla-hmGluR1b-GLAST in the β-lactamase assay and the Ca2+ mobilization assay. CHO-NFAT-bla-hmGluR1b and CHO-NFAT-bla-hmGluR1b-GLAST cells were plated and incubated with different concentrations of L-glutamate for 5 h, and then β-lactamase activity was determined. The results are shown as the blue/green ratio in (
To confirm the expression of GLAST in CHO-NFA- bla-hmGluR1b-GLAST, the GLAST mRNA level was determined using quantitative real-time PCR. GLAST mRNA levels normalized to GAPDH were 0.003 ± 0.00018 and 0.10 ± 0.012 (n = 3) in CHO-NFAT-bla-hmGluR1b and CHO-NFAT-bla-hmGluR1b-GLAST, respectively. The effect of a glutamate transporter inhibitor, trans-PDC, 21 on the β-lactamase assay in CHO-NFAT-bla-hmGluR1b-GLAST was evaluated to confirm that GLAST contributes to the improved S/B ratio in the assay. The basal activity of β-lactamase was 0.16 ± 0.0024 (n = 3) in the absence of trans-PDC, whereas the presence of 300 µM trans-PDC increased β-lactamase basal activity to 1.3 ± 0.19 (n = 3). L-glutamate still dose-dependently increased β-lactamase activity, with an EC50 value of 6.1 ± 1.7 µM in the presence of trans-PDC, whereas the S/B ratio was decreased to 2.1 ± 0.2 ( Fig. 5 ).
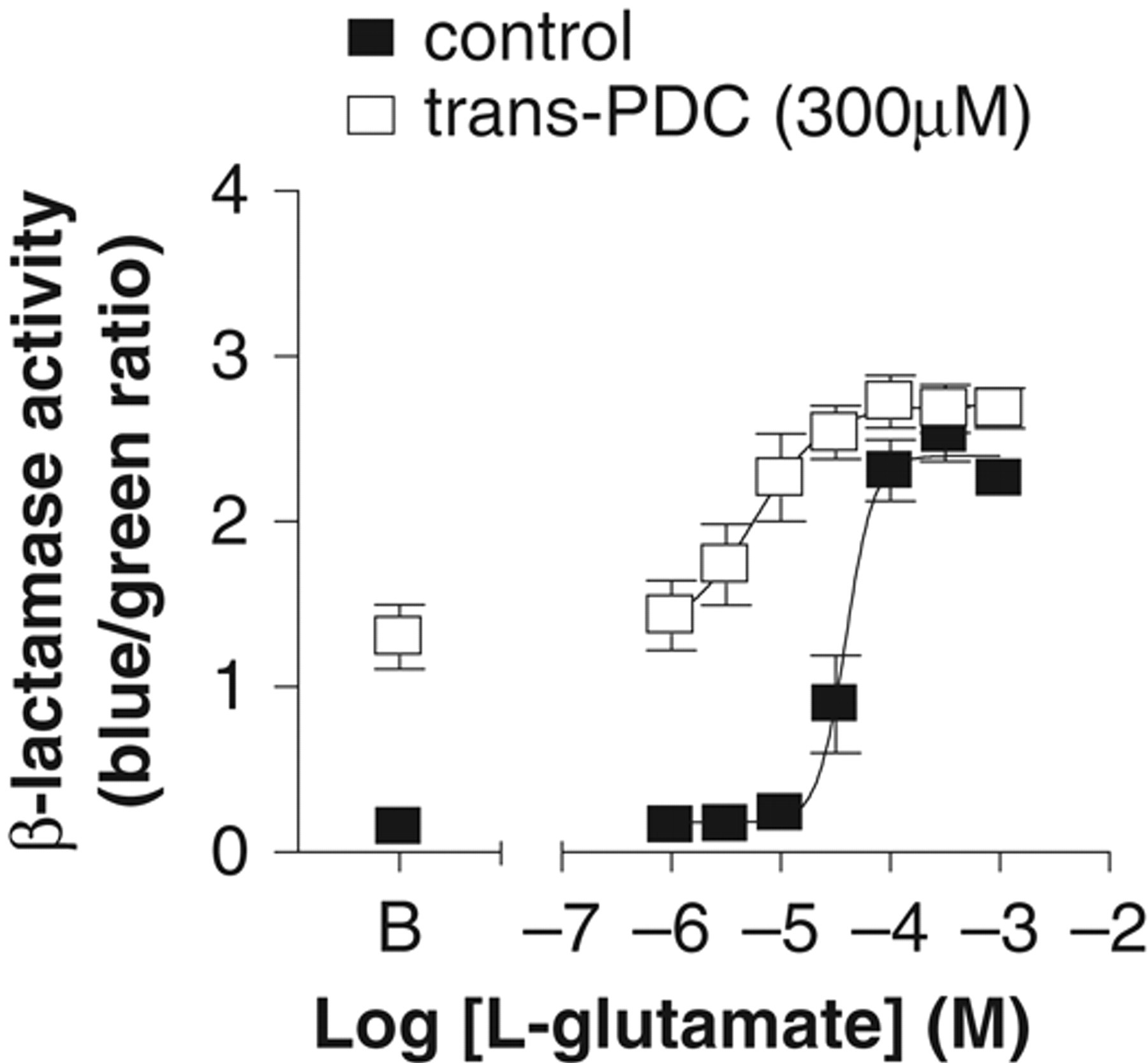
Effect of glutamate/aspartate transporter (GLAST) inhibitor, trans-PDC, on β-lactamase activity in CHO-NFAT-bla-hmGluR1b-GLAST. The cell line was plated and incubated with different concentrations of L-glutamate in the absence or presence of trans-PDC for 5 h, and then β-lactamase activity was determined. Closed and open squares represent β-lactamase activity in the absence or presence of 300 µM trans-PDC, respectively. “B” on the x-axis indicates basal β-lactamase activity in the absence of L-glutamate. The EC50 values of L-glutamate were 44 ± 10 and 6.1 ± 1.7 µM (n = 3) in the absence and presence of trans-PDC. Data are the means ± SEM from 3 individual experiments performed in duplicate.
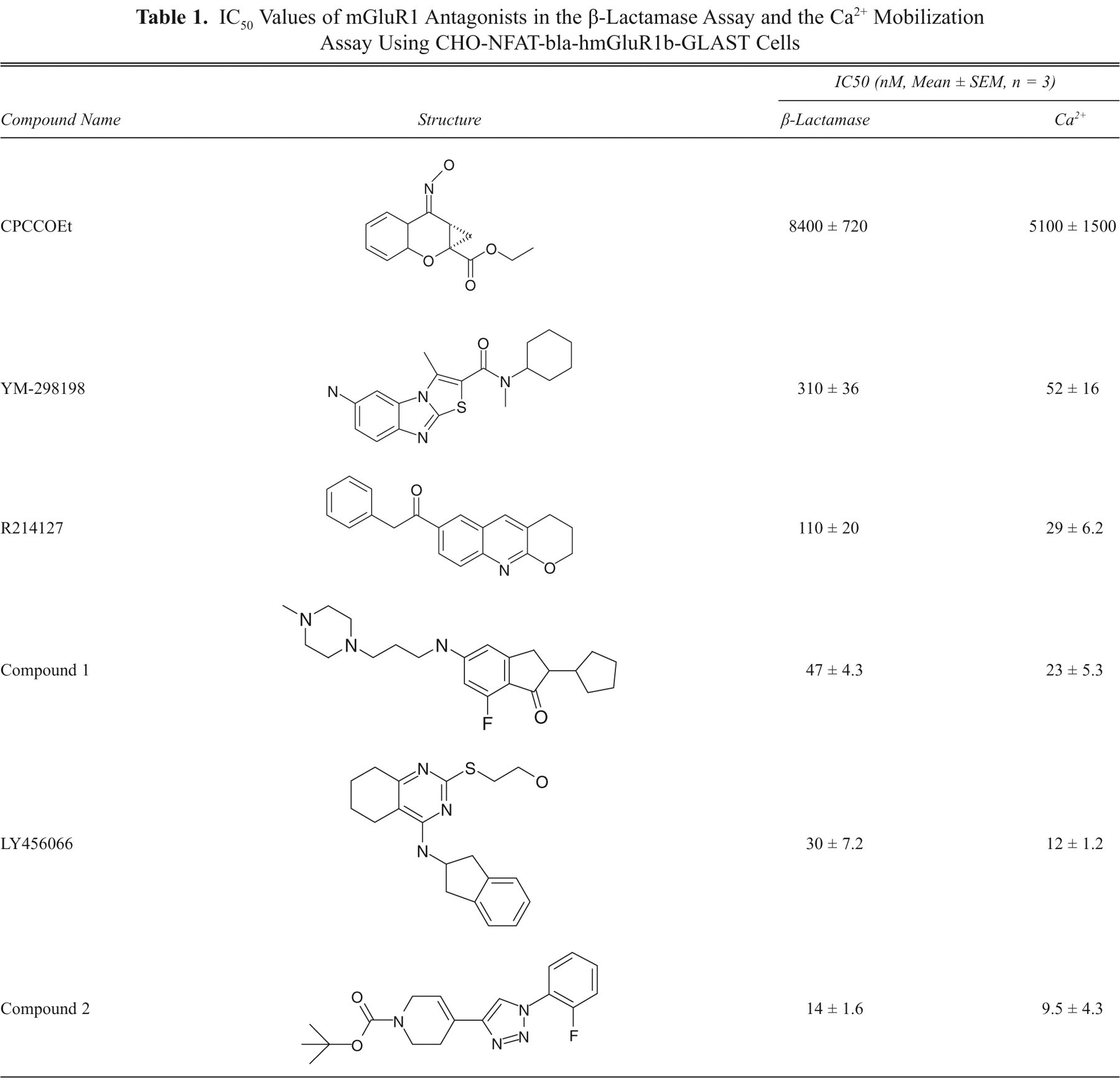
Inhibition of L-glutamate-induced β-lactamase activity by mGluR1 antagonists was validated and compared with that in a Ca2+ mobilization assay, using known mGluR1 antagonists from different structural classes 22-24 in addition to compound 1. All mGluR1 antagonists tested dose-dependently inhibited L-glutamate-induced β-lactamase activity, as well as intracellular Ca2+ increase elicited by L-glutamate ( Fig. 6A ; Table 1 ). Comparison of IC50 values of these antagonists showed good correlation (r 2 = 0.95) between the β-lactamase assay and the Ca2+ mobilization assay ( Fig. 6B ).
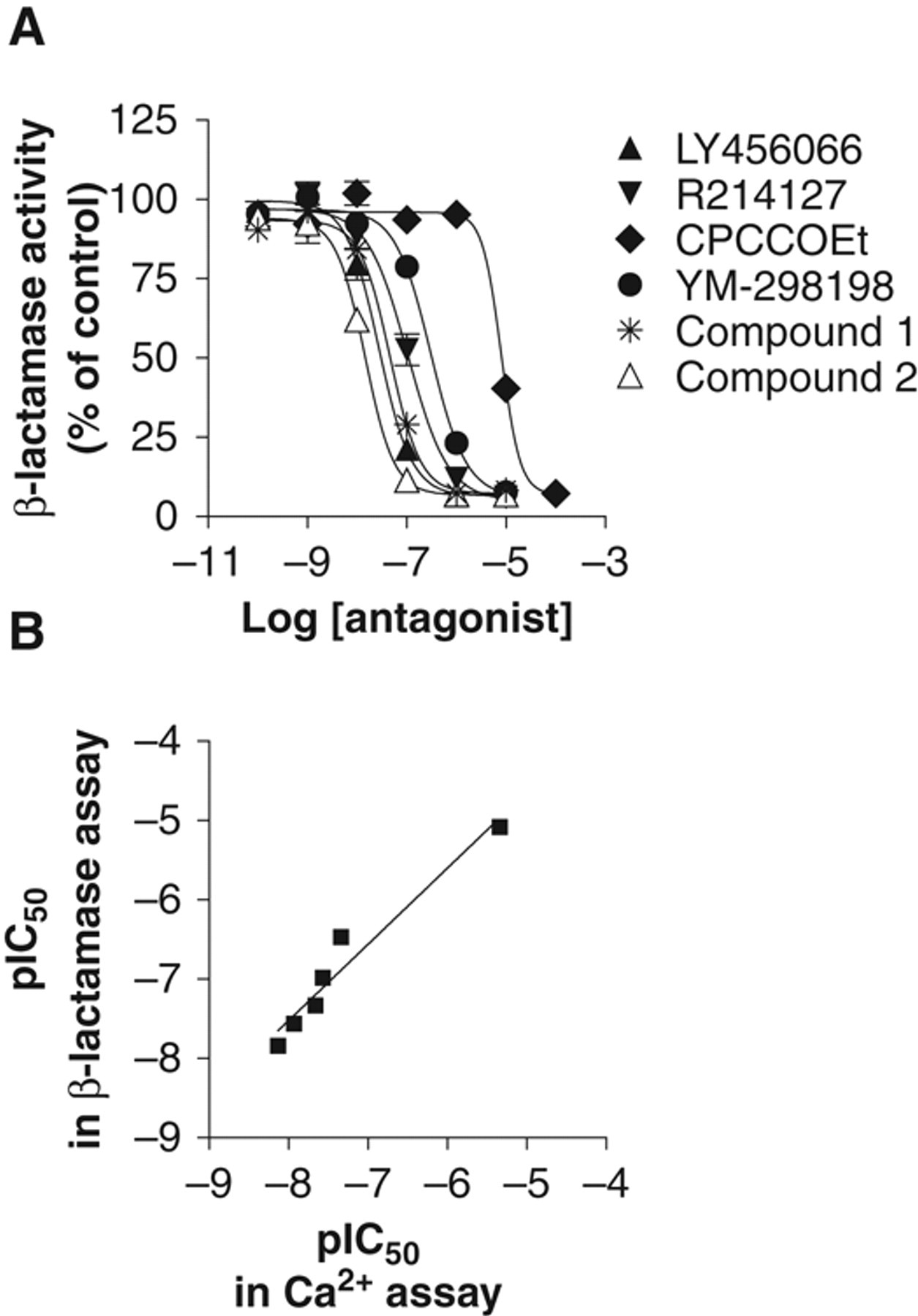
Evaluation of the mGluR1 β-lactamase assay and comparison with the mGluR1 Ca2+ mobilization assay using mGluR1 antagonists. (
IC50 Values of mGluR1 Antagonists in the β-Lactamase Assay and the Ca2+ Mobilization Assay Using CHO-NFAT-bla-hmGluR1b-GLAST Cells
Optimization of assay conditions
The optimum L-glutamate-induced β-lactamase response was determined in CHO-NFAT-bla-hmGluR1b-GLAST with respect to agonist stimulation time, incubation time with CCF2/AM, DMSO concentration, and stability of the cells and reagents. To determine the optimal agonist stimulation time, we incubated the cells at 37°C with L-glutamate for different times, and then CCF2/AM was added to determine β-lactamase activity. β-lactamase activity increased with increasing incubation time and reached a plateau level at 4 to 6 h ( Fig. 7A ). Optimum incubation time with CCF2 was determined by incubating the cells with the dye for different times after 5 h of agonist stimulation. As shown in Figure 7B , incubation with the dye for 2 to 4 h produced robust β-lactamase activity. Because test compounds in libraries are usually dissolved in DMSO, the effect of DMSO on L-glutamate-induced β-lactamase activity was also evaluated in CHO-NFAT-bla-hmGluR1b-GLAST. As shown in Figure 7C , 0.2% to 0.5% DMSO had no significant effect on L-glutamate-induced β-lactamase activity, whereas 1% DMSO slightly inhibited L-glutamate-induced β-lactamase activity. At 2% DMSO, β-lactamase activity induced by L-glutamate was significantly less than at the lower DMSO concentrations tested. In addition, β-lactamase assays were conducted with L-glutamate and cell suspensions prepared at different times because the stability of both L-glutamate and the cells is important for conducting high-throughput assays with an automated robotic system. The results indicated no significant differences in β-lactamase induction if L-glutamate and cell suspensions were freshly prepared or were 2 to 4 h old ( Fig. 7D ). Finally, using the optimized assay conditions, assay performance was examined across an entire plate. The representative S/B ratio and the coefficient of variation (CV) for the plate were 20 and 3.5%, respectively. The assay displayed a Z′ factor of 0.89.
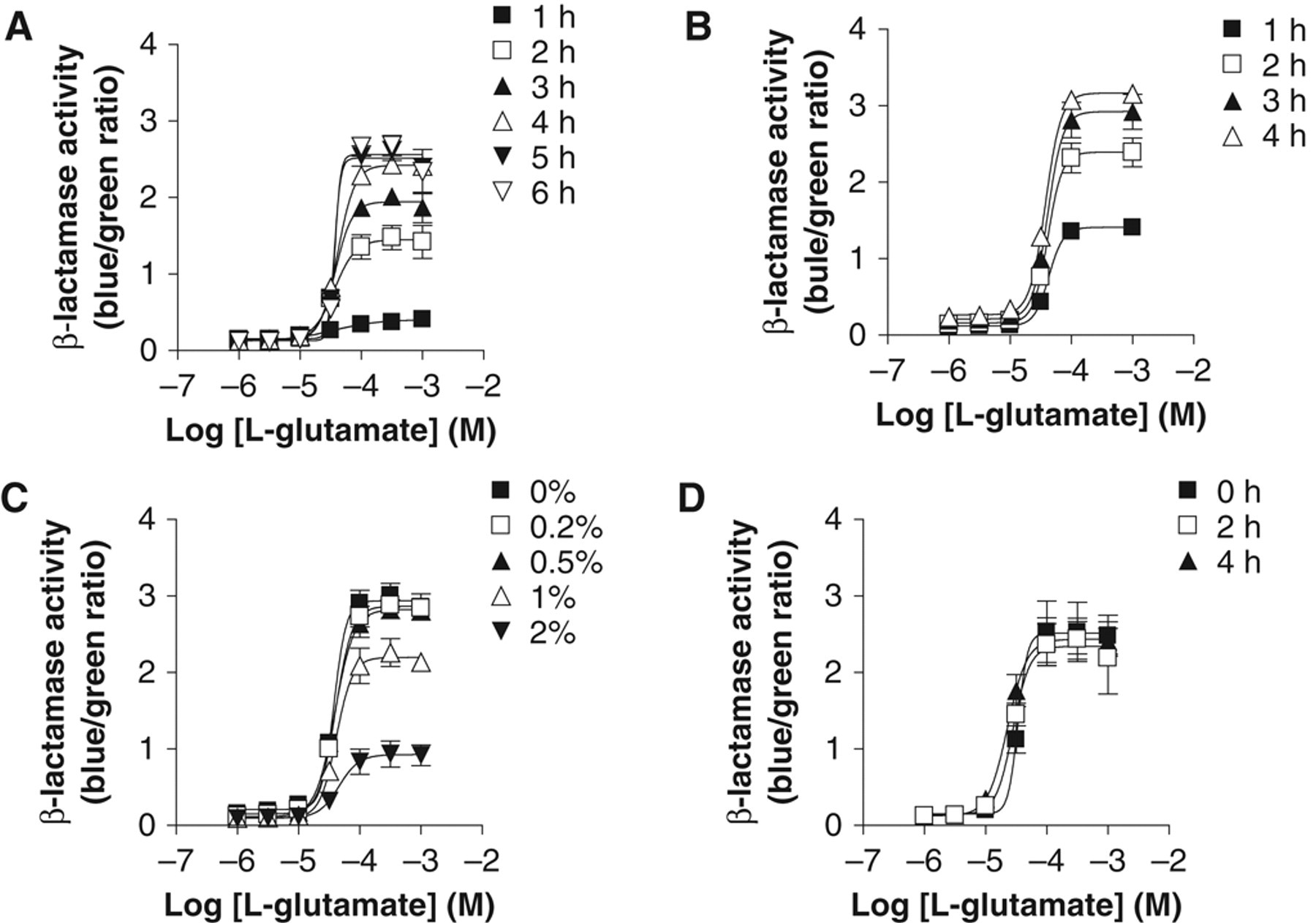
Optimization of the β-lactamase assay using CHO-NFAT-bla-hmGluR1b-GLAST for high-throughput screening. The optimum L-glutamate-induced β-lactamase response was determined in CHO-NFAT-bla-hmGluR1b-GLAST with respect to (
Discussion
In the present study, we developed and optimized a β-lactamase assay to identify mGluR1 antagonists. Coexpression of mGluR1 with GLAST in CHO-NFAT-bla cells was very effective for obtaining an S/B ratio sufficiently robust for HTS. This is the first report of a β-lactamase reporter gene assay effective for mGluR1.
Removal of L-glutamine from the assay medium decreased basal β-lactamase activity in CHO-NFAT-bla-hmGluR1b. L-glutamine is known to be easily converted into L-glutamate by chemical degradation. 25 Thus, high basal activity in the β-lactamase assay using CHO-NFAT-bla-hmGluR1b could be at least partially due to activation of mGluR1 mediated by L-glutamate generated from the degradation of L-glutamine. Even in the absence of L-glutamine, the mGluR1 antagonist, compound 1, inhibited β-lactamase activity below the basal level in CHO-NFAT-bla-hmGluR1b. This suggested that mGluR1 could still be activated in the absence of L-glutamine in the medium. One possible interpretation is agonist-independent constitutive activity of mGluR1. However, the CHO-NFAT-bla-hmGluR1b cells were expressing mGluR1b, which is a splice variant of mGluR1 with a smaller intracellular C-terminal domain and no constitutive activity. 26 Therefore, the reduction of basal β-lactamase activity in the presence of mGluR1 antagonist was less likely to be due to inhibition of constitutive activity by compound 1. An alternative interpretation could be that activation of mGluR1 was caused by endogenous L-glutamate secreted from CHO-NFAT-bla-hmGluR1b. CHO cells are known to release a significant amount of L-glutamate to activate mGluRs within a short period. 20 Release of endogenous L-glutamate may explain higher basal β-lactamase activity. Therefore, we developed a CHO-NFAT-bla-hmGluR1b-GLAST to reuptake newly released L-glutamate. As expected, the cell line showed lower basal activity and a greatly improved assay window in the β-lactamase assay compared with CHO-NFAT-bla-hmGluR1b.
High expression of GLAST mRNA in CHO-NFAT-bla-hmGluR1b-GLAST was confirmed by quantitative real-time PCR. A glutamate transporter inhibitor, trans-PDC, increased basal level β-lactamase activity and resulted in a lower S/B ratio in CHO-NFAT-bla-hmGluR1b-GLAST. These results supported the conclusion that coexpression of GLAST in CHO-NFAT-bla-hmGluR1b contributed to an improved S/B ratio in the β-lactamase assay. The result was also consistent with efficient mGluR1-mediated increase in phosphoinositide hydrolysis in cells coexpressing mGluR1 with GLAST. 27 In addition, the EC50 value of L-glutamate for the β-lactamase assay was potentiated in the presence of a GLAST inhibitor, trans-PDC, in CHO-NFAT-bla-hmGluR1b-GLAST. This result suggested that GLAST apparently lowered extracellular concentrations of L-glutamate, so more L-glutamate was required to activate the receptor. In agreement with this view, the EC50 value of L-glutamate in the presence of GLAST was higher than in the absence of GLAST in the β-lactamase assay. Also, the Hill coefficient in the presence of GLAST was higher than in the absence of GLAST, suggesting significant positive cooperativity in the presence of GLAST. The increased Hill coefficient might be due to direct or indirect interactions between mGluR1 and GLAST. However, as far as we are aware, such interactions have not been reported. Alternatively, the change may be accounted for by an increased population of mGluR1 homodimers in CHO-NFAT-bla-hmGluR1b-GLAST. A previous study using mutated receptors indicated that dimeric mGluR exhibited positive cooperativity in agonist-mediated functional response. 28 The ability of GLAST to lower extracellular L-glutamate concentrations might cause a blockade of agonist-induced mGluR1 internalization and an increased ratio of mGluR1 dimers on the cell surface, resulting in increased positive cooperativity and increased Hill coefficients. Further studies will be necessary to validate this hypothesis.
To evaluate the utility of the β-lactamase assay for the identification of mGluR1 antagonists, inhibitory activities of structurally diverse mGluR1 antagonists were tested in the β-lactamase assay and compared with those in the Ca2+ mobilization assay. All tested compounds inhibited L-glutamate-induced β-lactamase activity in a dose-dependent manner. The rank order of potency in the β-lactamase assay was similar to that in the Ca2+ mobilization assay. The IC50 values of the compounds obtained by the 2 assays correlated well, although the values in the β-lactamase assay were somewhat higher than those in the Ca2+ mobilization assay. The slight shift of the IC50 values in the β-lactamase assay might be due to more sensitive detection of agonist response in the reporter gene assay, where receptor-mediated intracellular signals such as Ca2+ can be amplified. These results suggest that the β-lactamase assay using CHO-NFAT-bla-hmGluR1b-GLAST could be applied to HTS for the identification of mGluR1 antagonists.
The present study describes an alternate approach for identifying mGluR1 ligands by functional screening. Some antagonists of gonadotropin-releasing hormone receptor were found to be much more potent in the β-lactamase assay than in the Ca2+ mobilization assay. 15 This finding suggests that mGluR1 antagonist screening using the β-lactamase assay might be useful in identifying novel compounds not detected by the conventional Ca2+ mobilization assay. In addition, as mentioned above, the β-lactamase assay could be more sensitive than the Ca2+ mobilization assay. Thus, if this assay is applied in agonist screens, it will allow more sensitive detection of compounds with low potency and/or weak intrinsic activity (such as partial agonists) compared with second-messenger assays. On the other hand, the β-lactamase assay is based on an increase in intracellular Ca2+ level, activation of calcineurin, translocation of NFAT, and the resultant induction of β-lactamase activity. It is therefore possible that a compound that affects the signaling pathway, such as a calcineurin inhibitor, will give false positives in mGluR1 antagonist screening. Therefore, a NFAT-based β-lactamase assay for another Gq-coupled receptor will be useful for eliminating such compounds. Optimization of the assay conditions showed that appropriate times for agonist stimulation and CCF2/AM loading were 5 and 2 h, respectively. The cell suspension and L-glutamate solution were stable, with no significant differences observed between freshly prepared and 4-h-old preparations; 0.5% DMSO was tolerated in the assay. A Z′ factor can be used for quality assessment in assay development and optimization. A Z′ factor higher than 0.5 is indicative of a sensitive assay with low variation in replicates and is therefore often used as the criterion for a robust HTS assay. 19 In the present study, the Z′ factor for the β-lactamase assay using CHO-NFAT-bla-hmGluR1b-GLAST was 0.89, thus satisfying this criterion and suggesting that it is a reliable assay.
As mentioned above, this is the first description of a β-lactamase reporter assay among mGluR subtypes, although a cAMP-responsive luciferase assay for mGluR7 has previously been reported. 29 In the luciferase assay for Gi-coupled mGluR7, activation of the receptor inhibited forskolin-stimulated luciferase activity, without using a glutamate transporter such as GLAST. This might be explained by a unique characteristic of mGluR7—namely, that it has very low potency toward L-glutamate. 30 Thus, the concentration of L-glutamate released from the cells might be insufficient to activate mGluR7. The present finding that GLAST coexpression improved the sensitivity of the β-lactamase assay for mGluR1 might be useful for developing reporter gene assays not only for mGluR1 but also for other mGluR subtypes (excluding mGluR7).
In conclusion, a β-lactamase-based assay for mGluR1 was developed and optimized in the present study. We demonstrated that coexpression of mGluR1 with GLAST in CHO-NFAT-bla cells was useful for achieving lower background and improved S/B ratio in the β-lactamase response. The β-lactamase assay for mGluR1 is comparable to the Ca2+ mobilization assay in evaluating mGluR1 antagonists from different structural classes, suggesting that the β-lactamase assay is applicable to ligand screening for mGluR1. Success in developing the β-lactamase assay for mGluR1 was mainly due to GLAST coexpression. Utilization of GLAST may be helpful for developing reporter gene assays for other mGluR subtypes. The β-lactamase reporter assay with CHO-NFAT-bla-hmGluR1b-GLAST should be amenable to a high-throughput format for mGluR1 antagonist screening.
Footnotes
Acknowledgements
The authors thank Professor Kazuhide Inoue (Kyushu University) for supporting the preparation of this article, and Dr. Naohiro Tsukamoto for technical support and useful discussions.