
Abstract
KCC2, potassium chloride cotransporter 2, is expressed exclusively in the CNS (on inhibitory neurons) and plays a major role in maintaining appropriately low intracellular chloride levels that ensure inhibitory actions of GABAA and glycine receptors. As such, it plays a pivotal role in inhibitory mechanisms that control neuronal excitation in the CNS. KCC2 downregulation has been implicated in various excitatory disorders, such as epilepsy and neuropathic pain. Positive modulators of KCC2 expression or activity may thus provide effective therapy for these disorders. However, the identification of such agents is hindered by the lack of a high-throughput screening method. Here the authors report the development of a fluorescence-based thallium (Tl+) transport assay using a Fluorometric Imaging Plate Reader (FLIPR), in which KCC2 activity is assessed by measuring the initial rate of KCC2-mediated Tl+ transport/influx. The authors demonstrate Tl+/Cl− cotransport by KCC2, which exhibits a high apparent affinity for Tl+ and dependency on the presence of the Cl− ion. Pharmacological studies revealed anticipated effects and potencies of known KCC-positive (NEM, staurosporine) and KCC-negative (DIOA, furosemide) modulators. The authors demonstrate that the assay is robust and reproducible and can be employed in high-throughput screening for positive modulators of KCC2 as potential therapeutic agents.
Introduction
T
Multiple lines of evidence suggest that the expression level and activity of KCC2 are critical in neuronal Cl− homeostasis. 3 KCC2 can be downregulated with increased neuronal activity—for example, in epilepsy or with nerve injury in various pathological conditions, such as with neuropathic pain and after trauma. The change in KCC2 activity leads to increased intracellular Cl− concentration. 4,5 As a result, Cl− influx upon activation of GABAA or glycine receptors can be reduced or even reversed, thus reducing inhibition and potentially shifting the receptor-mediated responses from hyperpolarizing inhibition to depolarizing excitation. 6
The important role of KCC2 in inhibitory responses of GABA and glycine has been supported by data from genetic and pharmacological studies. Mice deficient in KCC2 exhibit frequent generalized seizures and die shortly after birth. The heterozygotes, although appearing normal, show increased susceptibility to epileptic seizures. 7,8 In pharmacological studies, administration of [(dihydroindenyl)oxy] alkanoic acid (DIOA), a KCC-selective inhibitor, to the spinal cord increases lamina I responses to noxious and innocuous stimuli and causes spontaneous burst firing of neurons, mirroring symptoms of neuropathic pain. 9 In addition, intrathecal blockade (with DIOA) or knockdown (with KCC2-specific antisense) of KCC2 causes significant allodynia and hyperalgesia in naïve rats. 10 Taken together, these findings strongly suggest that downregulation of KCC2, and hence disruption of anion homeostasis, contributes to increased neuronal excitability in various pathological states and that a positive modulator of KCC2 expression or activity may provide effective therapy for epilepsy or neuropathic pain.
In vitro studies have revealed that KCC2 activity can be upregulated by several small-molecule agents, including N-ethylmaleimide (NEM), a cysteine-alkylating agent that can modify proteins by reacting with sulfhydryls, and staurosporine, a broad-spectrum kinase inhibitor. 11,12 However, the effects of these agents on KCC2 are likely via indirect mechanisms. 12,13 To our knowledge, no positive modulator has been published to date that acts directly and specifically on KCC2. Several functional assays have been reported to assess KCC2 activity, including a patch-clamp assay to measure the glycine reversal potential driven by KCC2, 14 an assay measuring radioactive rubidium (86Rb+) transport via KCC2, 11 and a KCC2-mediated NH4 + uptake assay measured with a pH-sensitive fluorescent dye. 15 However, these assays suffer from low throughput and poor temporal resolution, sensitivity, or ease of use and are not amenable to high-throughput screening (HTS) for KCC2 modulators.
In this report, we describe the development of a fluorescence-based thallium (Tl+) influx assay that can accurately and rapidly measure KCC2 activity in mammalian cells. This assay can be employed to screen for positive modulators of KCC2 from large chemical libraries.
Materials and Methods
Reagents
BTC-AM dye was obtained from Invitrogen (Carlsbad, CA). All chemicals were purchased from Sigma (St. Louis, MO) except NEM from Pierce (Rockford, IL).
Cloning of human KCC2
To clone human KCC2 (hKCC2, GenBank accession number: NM_020708) for expression, the coding sequence of a full-length cDNA clone obtained from OriGene (Rockville, MD) was amplified by PCR, sequence confirmed, and cloned into the pcDNA3.1/V5-his TOPO expression vector (Invitrogen).
Transfection and cell culture
Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Invitrogen) in a humidified 5% CO2, 95% O2 incubator at 37°C. Cells were transfected with the hKCC2 expression construct using LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s instructions.
To generate a HEK293 cell line stably expressing human KCC2, the hKCC2 expression construct was linearized by SalI (Invitrogen) and transfected into HEK293 cells. Stable cell lines were generated by antibiotic selection (1 mg/mL Geneticin, Invitrogen) 48 h posttransfection. Single-cell colonies were selected 14 days posttransfection and amplified, and the expression of hKCC2 was assessed by Western blot analysis.
Western blot
To detect the expression of KCC2 in HEK293 cells, whole-cell lysates were prepared by lysing cells in RIPA buffer (1× phosphate-buffered saline [PBS], 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) and subjected to Western blot analysis with an antibody raised against KCC2 (Millipore, Billerica, MA).
Tl+ influx FLIPR assay
Cells were seeded in 96-well, black-walled, clear-bottomed, poly-D-lysine-coated plates (BD Biosciences, Bedford, MA) at a density of 5 × 104 cells per well 24 h before the assay. On the assay day, BTC-AM dye was loaded into the cells by replacing the cell culture medium with 100 µL/well of 4 µM dye in DPBS (Invitrogen). Dye loading was allowed to proceed for 2 h at room temperature, and then cells were washed twice in 100 µL/well of assay buffer (in mM: 10 HEPES 7.3, 10 glucose, 1 NaH2PO4, 1 CaCl2, 1 MgSO4, 140 NaCl, 0.1 Ouabain, 0.01 bumetanide) to remove unloaded dye. The wash buffer was discarded, and the cells were incubated in 50 µL of assay buffer before loading onto a Fluorometric Imaging Plate Reader (FLIPR) system (Molecular Devices, Sunnyvale, CA). Compounds to be assayed were added to the cells in 50 µL of assay buffer and incubated for 4 min. The FLIPR signal was initiated by adding 100 µL of assay buffer containing 2 mM TlNO3 (Sigma). Fluors were excited using the 488-nm line of an argon laser, and emission was filtered using a 540 ± 30-nm bandpass filter. Fluorescent signals were recorded for 5 min, and the initial rate of Tl+ transport was deduced by analyzing the linear increase of fluorescent signals within the initial 10 s following Tl+ addition.
Data analysis
Relative Tl+ transport initial rates were calculated as a percentage of the control and plotted against external ion concentrations. Data were collected from at least 6 trials at each concentration and fit with a single-site binding curve using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA). Michaelis constant Km was deduced. Relative KCC2 activities in the presence of inhibitors were calculated as a percentage of the control. Data were collected from 4 trials at each inhibitor concentration and fit with a nonlinear regression curve using GraphPad Prism. IC50 values were calculated from the resulting sigmoidal dose-response curves. Z′ factor was calculated according to Zhang et al. 16 using the following equation: Z′ = 1 – [3 SD of sample + 3 SD of control]/[mean of sample – mean of control]. Data are presented as mean ± SEM.
Results
Development of a KCC2-mediated Tl+ influx assay
86Rb influx assay has been widely adopted to measure the activity of KCC/NKCC cotransporters, including KCC2. 11,17,18 We sought to develop a similar assay using Tl+ as the indicator ion instead of the radioactive 86Rb. This became potentially feasible owing to the availability of a Tl+-sensitive, fluorescence-based ion flux indicator (BTC-AM) and the successful use of this dye in a high-throughput assay for the identification and characterization of potassium channel modulators. 19 Many cations can be transported by KCC2, including K+, Rb+, NH4 +, and Cs+, 15 and we hypothesized that Tl+ may also be transportable. KCC2, as an ion transporter, has bidirectional transport ability and cotransports ions into or out of the cell depending on existing ionic gradients. Tl+ transported by KCC2 may associate with the BTC-AM dye, causing a fluorescence change that can be detected by FLIPR. Hence, the influx of Tl+ as detected in this assay could be employed to measure KCC2 activity.
We sought to assess the feasibility of this assay in HEK293 cells using heterologous expression of recombinant human KCC2. In preliminary experiments, however, a fairly robust Tl+ influx signal was observed in the parental HEK293 cells (data not shown). This background signal is most likely attributable to an endogenous NKCC-like sodium potassium chloride cotransporter activity reported to exist in HEK293 cells and can be blocked by bumetanide. 11,20 Indeed, when HEK293 cells were pretreated with 10 µM bumetanide (added in the assay buffer), the background Tl+ influx/transport signal was significantly reduced (data not shown). It has been reported by several groups that bumetanide is a potent NKCC blocker, whereas at 10 µM, its inhibition of KCC2 is minimal. 11,21,22 Therefore, the use of this concentration of bumetanide should not interfere with KCC2 activity. Thus, with sufficient block of endogenous ion transporter activity in HEK293 cells, KCC2-driven Tl+ transport could be specifically measured.
We cloned the human KCC2 and transiently transfected HEK293 cells with different amounts of KCC2 expression construct. The expression of hKCC2 was evaluated by Western blot analysis using an anti-KCC2 antibody. A specific ~140-kDa protein band, which represents the monomer form, and multiple protein bands above 200 kDa, which likely represent the reported oligomeric forms of KCC2, were detected ( Fig. 1A ). 23 In the Tl+ influx FLIPR assay, robust Tl+ transport signals were observed in HEK293 cells transfected with hKCC2 in a dose-dependent manner but not in mock-transfected HEK293 cells from which only a background signal was observed ( Fig. 1B ). The KCC2-driven FLIPR signal displays a rapid linear increased phase within the first 10 s after Tl+ addition followed by a slower increase and eventually a plateau phase. Kinetic measurement of transporter activity follows the same fundamental concepts and principles as measurement of enzyme activity. Thus, KCC2 activity is most accurately measured by the initial rate of Tl+ transport, which can be calculated using the initial linear phase of signal increase. We observed a substantial increase in the initial rate of Tl+ transport into KCC2-transfected cells in a dose-dependent manner over the mock-transfected cells, suggesting that the increase is driven by KCC2 expression ( Fig. 1C ).
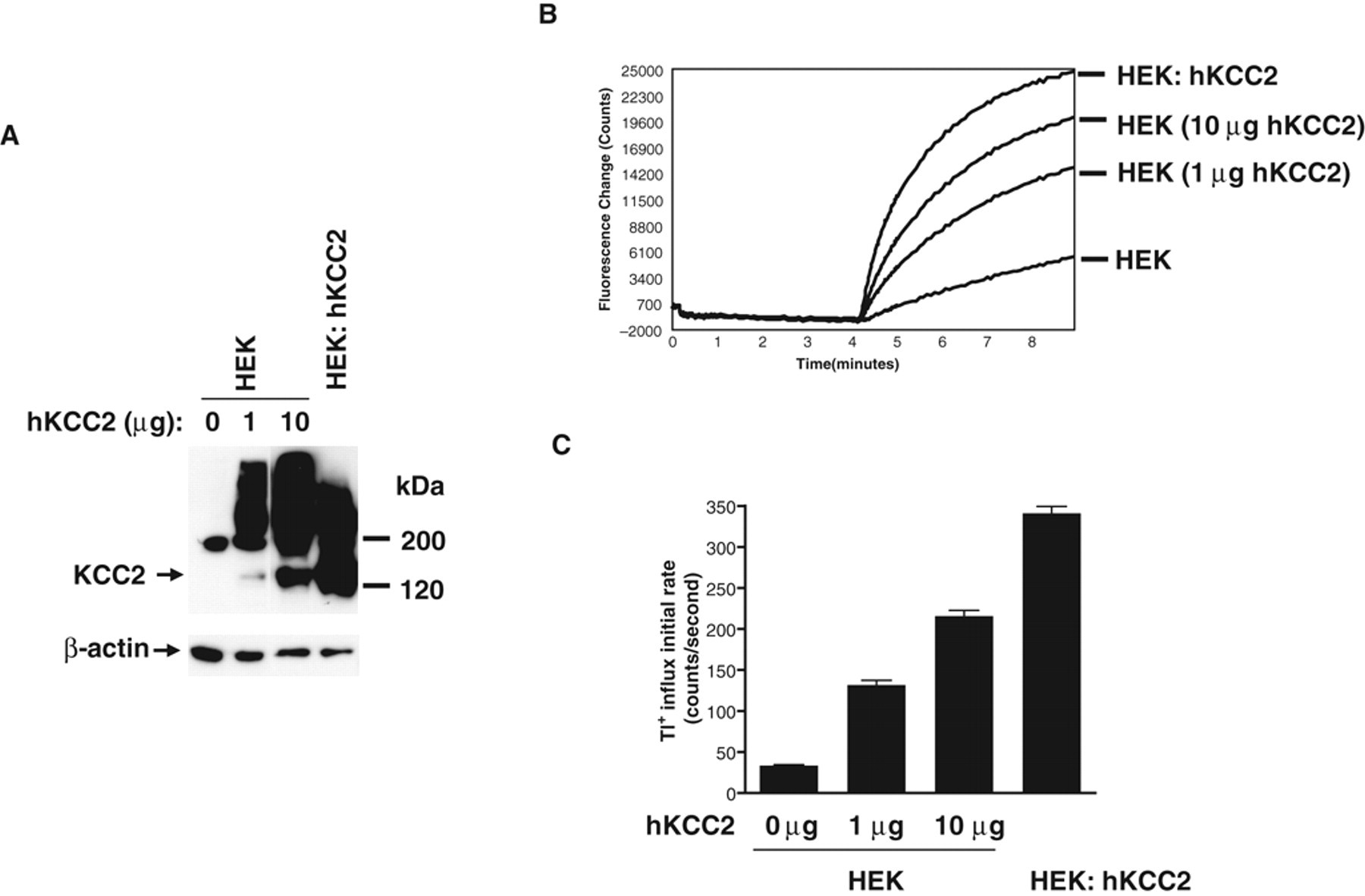
Tl+ influx assay for human KCC2 (hKCC2) activity in HEK293 cells. (
To identify positive modulators for KCC2, a HEK293 cell line stably expressing human KCC2 (HEK: hKCC2) was established. In this cell line, KCC2 was expressed even more robustly than by transient transfection, and as a result, we observed a higher initial rate of KCC2-driven Tl+ transport in our FLIPR assay ( Fig. 1 ). The initial rate of Tl+ influx observed in the HEK: hKCC2 cell line was approximately 10-fold over the background signal in parental HEK293 cells.
External ion dependency of the KCC2-mediated Tl+ transport
To assess the dependency of the KCC2-mediated Tl+ transport on external Cl− concentration, assay buffers with different concentrations (0-140 mM) of Cl− were prepared in which NaCl was replaced by Na-gluconate to maintain appropriate osmolarity. The cells were quickly washed twice in these assay buffers prior to the FLIPR assay. As expected for a K-Cl cotransporter, KCC2-mediated Tl+ transport requires external Cl−, and the initial rate of transport increases with increasing concentration of Cl− ( Fig. 2A ). A calculation of the affinity of hKCC2 for external Cl− revealed a Michaelis constant Km at 67.3 ± 3.9 mM (n = 8), in line with that determined by the 86Rb influx assay. 11 Thus, in the final optimized format of the Tl+ transport assay, external Cl− approximating the physiological concentration of 140 mM was applied, a concentration that ensures that Cl− ion is not a limiting factor in measuring KCC2 activity.
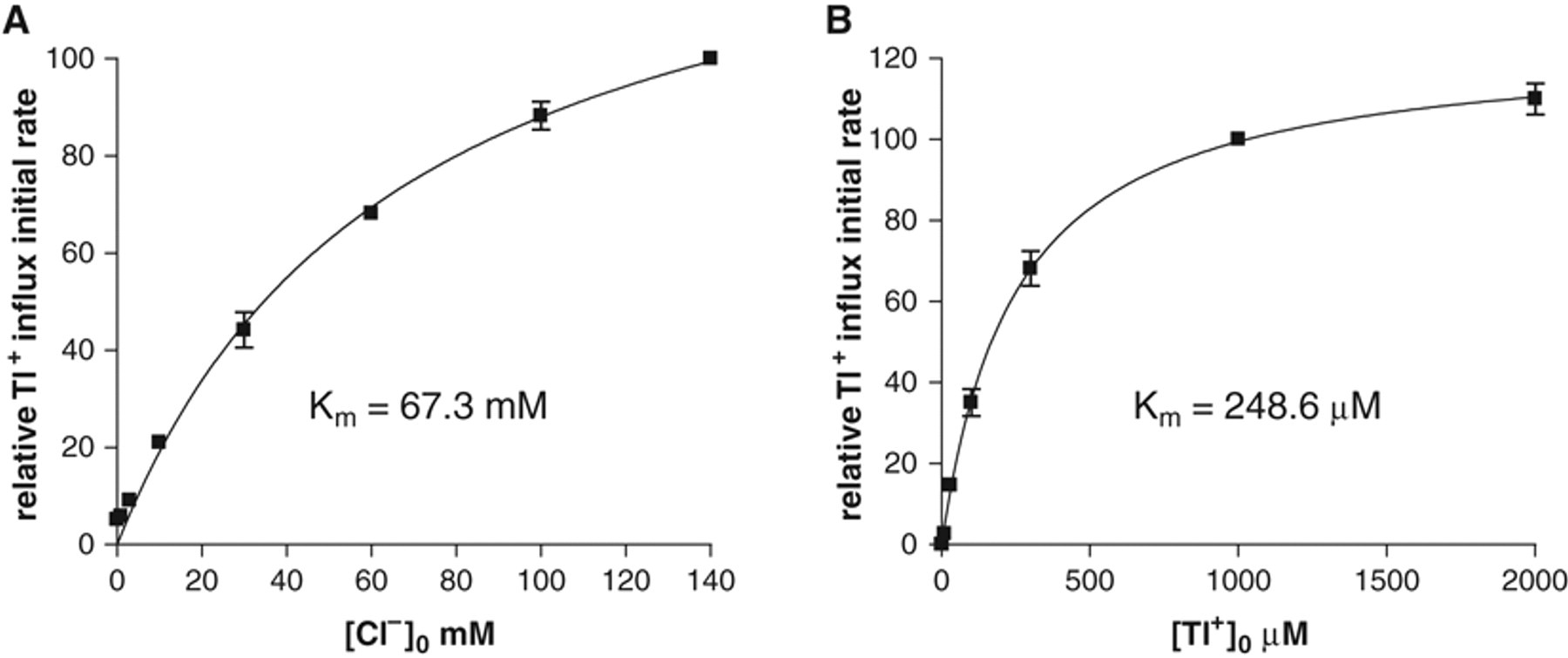
Dependency of the initial rate of KCC2-mediated Tl+ influx on external Cl− and Tl+. (
The dependency of the KCC2-mediated Tl+ transport on external Tl+ concentration was also assessed. Different concentrations of Tl+ were employed to initiate the influx signal, and the relative initial rates of influx were plotted against Tl+ concentration ( Fig. 2B ). KCC2-driven Tl+ influx exhibited a high affinity for external Tl+ with a Michaelis constant Km at 248.6 ± 17.9 µM (n = 6). In the final optimized assay format, we use 1 mM Tl+ to initiate KCC2-mediated Tl+ transport, a concentration high enough to ensure that the Tl+ ion is not limiting and that the Tl+ transport signal reflects KCC2 activity. To assess whether Tl+ influx can be competed by external K+, assay buffers were made with increasing concentrations of K+ by replacing NaCl with KCl and the Tl+ influx initiated by 1 mM Tl+. No significant effect on the Tl+ transport signal was observed by addition of external K+ up to 10 mM, whereas a higher concentration of added K+ (above 30 mM) inhibited the Tl+ transport signal in a concentration-dependent manner. The inhibition by K+ suggests that Tl+ and K+ can compete for transport by KCC2, providing further evidence of the specificity of transport of Tl+ by KCC2.
Inhibition of Tl+ transport by KCC-negative modulators
To evaluate whether the Tl+ influx assay can be employed to measure modulators of KCC2 activity, we assessed the effects of known inhibitors of KCC2. The alkanoic acid DIOA has been reported to be a potent and selective inhibitor of K-Cl cotransporters. 24 We observed that DIOA inhibits the initial rate of KCC2-mediated Tl+ influx in a concentration-dependent manner, with an IC50 value of 13.4 µM (n = 4; Fig. 3A ). This value is consistent with the report that 30 µM DIOA treatment blocked KCC2 activity in spinal cord lamina I neurons. 10
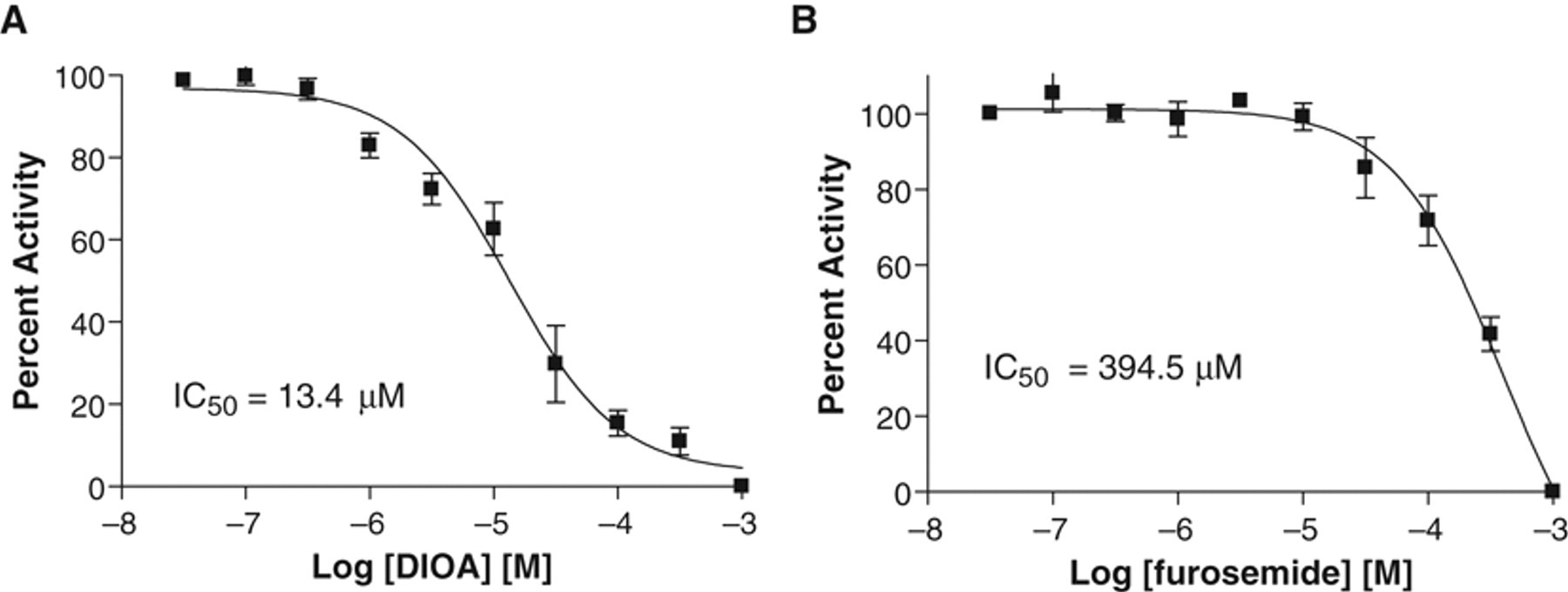
Inhibition of KCC2 activity by [(dihydroindenyl)oxy] alkanoic acid (DIOA) and furosemide determined by the Tl+ influx assay. (
The loop diuretic furosemide is a general NKCC and KCC blocker. 1 Furosemide also inhibits the initial rate of KCC2-driven Tl+ influx in a concentration-dependent manner, albeit with significantly less potency (IC50 = 394.5 µM; n = 4) than DIOA ( Fig. 3B ), as has also been reported by others. 1
Stimulation of Tl+ transport by KCC-positive modulators
We further validated the Tl+ influx assay by assessing positive modulators of KCC2 activity. NEM has been reported to stimulate KCC2 activity in various assay systems. 1 Treatment of HEK: hKCC2 cells with NEM for 15 min resulted in a concentration-dependent increase in the initial rate of KCC2-mediated Tl+ influx ( Fig. 4 ). At 100 µM, NEM stimulated KCC2 activity by approximately 3-fold.
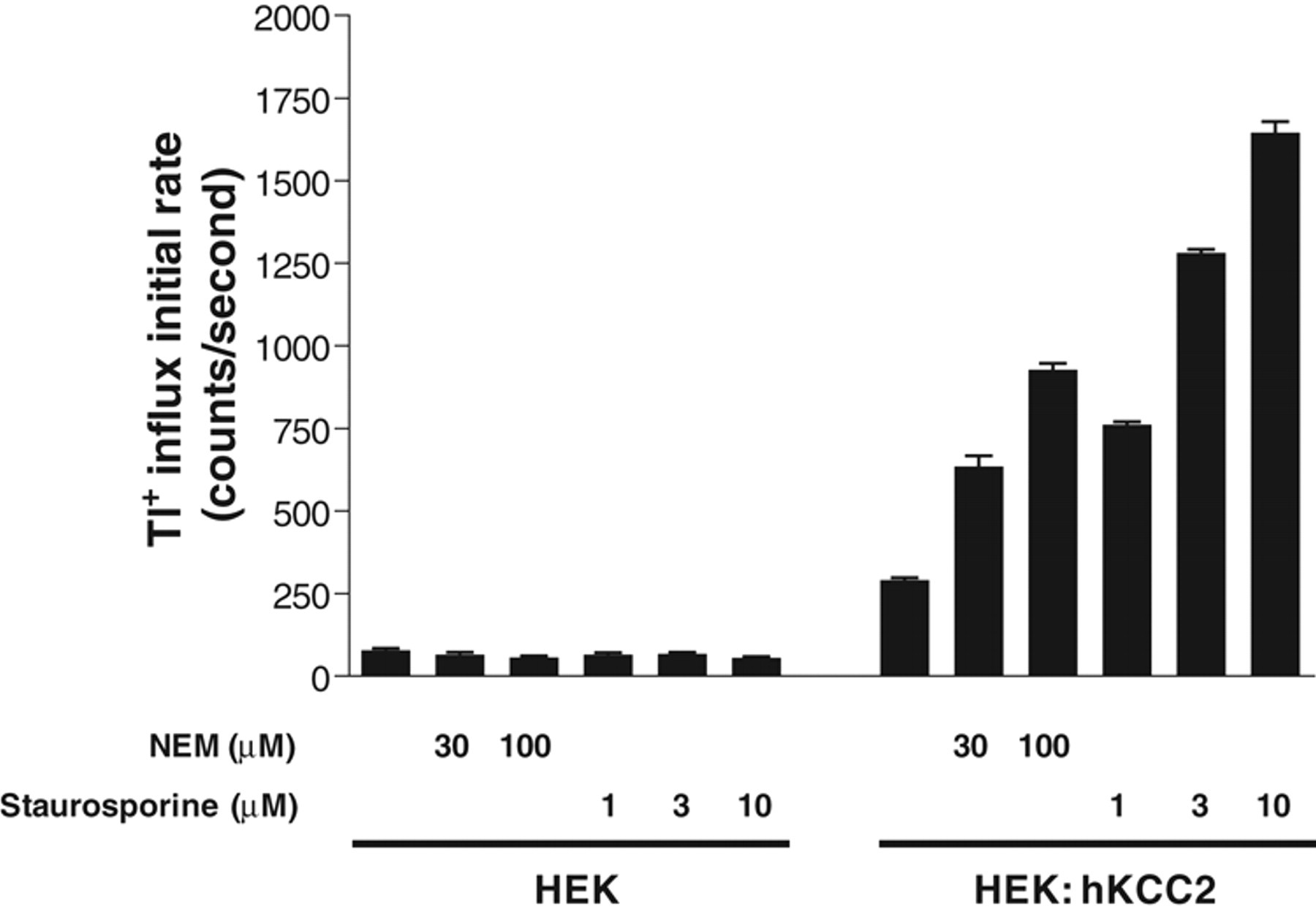
Stimulation of KCC2 activity by positive modulators N-ethylmaleimide (NEM) and staurosporine determined by the Tl+ influx assay. HEK293 cells (HEK) or KCC2 stable cell line (HEK: hKCC2) were treated with different concentrations of NEM or staurosporine, as shown, for 15 min prior to the addition of Tl+. The initial rates of Tl+ influx under these treatments versus that in untreated cells are shown (n = 16).
We also assessed staurosporine, which has been reported to be a positive modulator of KCC2 activity. 12,25 As expected, treatment of HEK: hKCC2 cells with staurosporine for 15 min resulted in a concentration-dependent stimulation of the initial rate of KCC2-mediated Tl+ influx ( Fig. 4 ). The stimulation by staurosporine is more significant than that by NEM. At 10 µM, staurosporine stimulated KCC2 activity greater than 5-fold.
To confirm that the stimulatory effects of NEM and staurosporine were mediated by KCC2, the effects of the 2 modulators on the background Tl+ influx signal in HEK293 cells was also assessed. Neither NEM nor staurosporine treatment significantly affects the background Tl+ influx in HEK293 cells ( Fig. 4 ), suggesting that NEM and staurosporine stimulate Tl+ influx by modifying (directly or indirectly) KCC2. The observed stimulatory effects of NEM and staurosporine on KCC2 activity are consistent with previous reports and also indicate that novel positive modulators of KCC2 activity can be identified using this assay.
Assay performance for HTS
To assess whether the Tl+ influx assay is suitable for HTS, we determined the Z′ factor, a statistical parameter reflecting the quality of an HTS assay by comparing the assay dynamic range to data variation. 16 The test was conducted using 5 experimental plates on which cells on half of the plate were treated with 10 µM staurosporine as positive control and the other half with no compound as negative control ( Fig. 5 ). The value of Z′ factor thus calculated ranged from 0.63 to 0.74 with an average of 0.68 ± 0.019 (n = 5). In general, an assay with a Z′ factor greater than 0.5 is suitable for HTS. Our results demonstrate that the Tl+ influx assay is robust and reproducible.
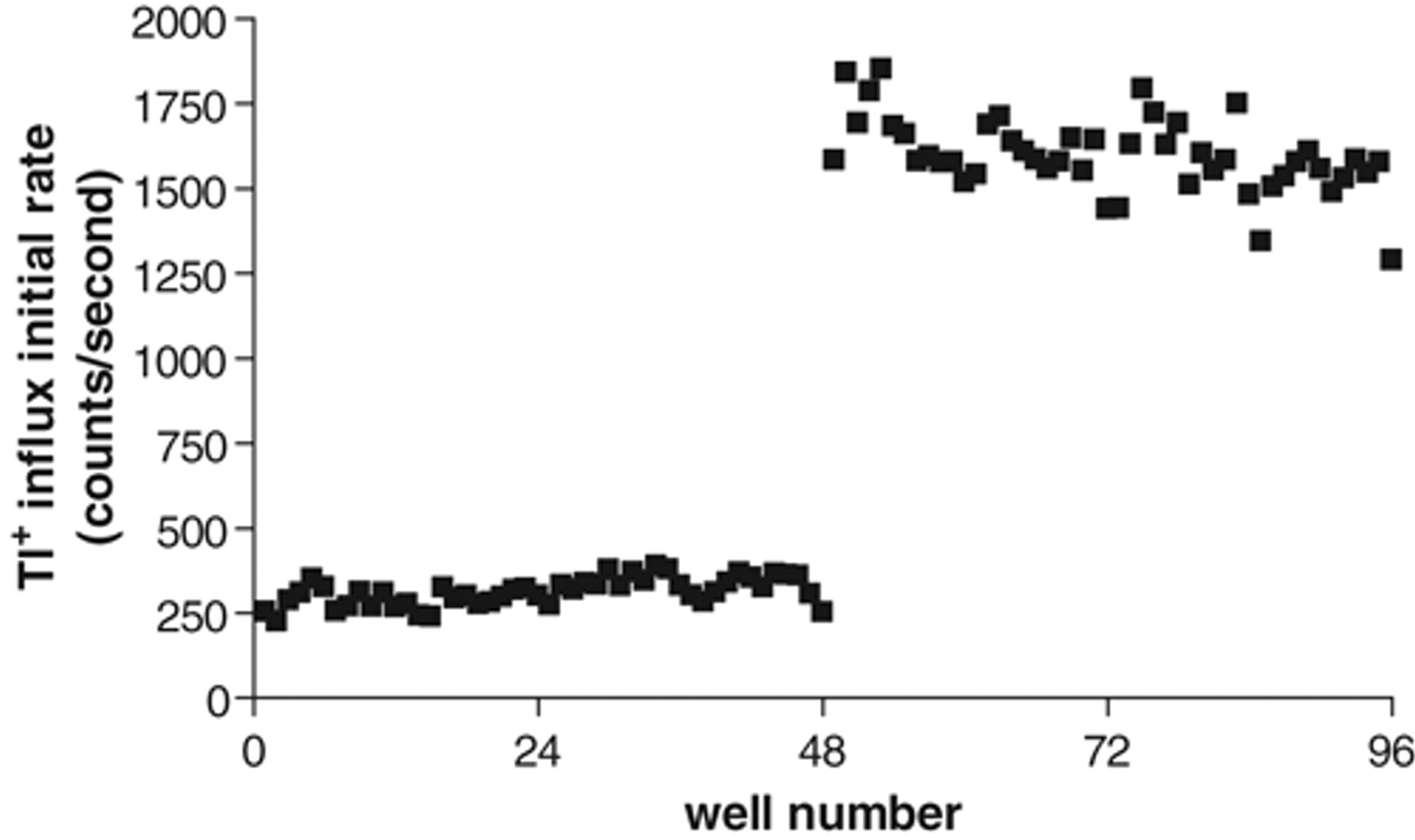
Scatter plot of Tl+ influx signals from an example test plate on which cells on half of the plate were treated with 10 µM staurosporine as positive control and the other half with no compound as negative control. The average Z′ factor calculated from 5 such assays was 0.68.
During HTS, DMSO is routinely used to solubilize compounds so the effect of DMSO on KCC2-mediated Tl+ influx was evaluated. DMSO was tested at final concentrations ranging from 0.25% to 5% (v/v). Increasing the concentration of DMSO from 1% to 5% produced a concentration-dependent decrease in KCC2-mediated Tl+ influx FLIPR signal, whereas minimal interference was observed at 0.5% DMSO ( Fig. 6 ). Therefore, DMSO concentration should be maintained at no more than 0.5% during screening.
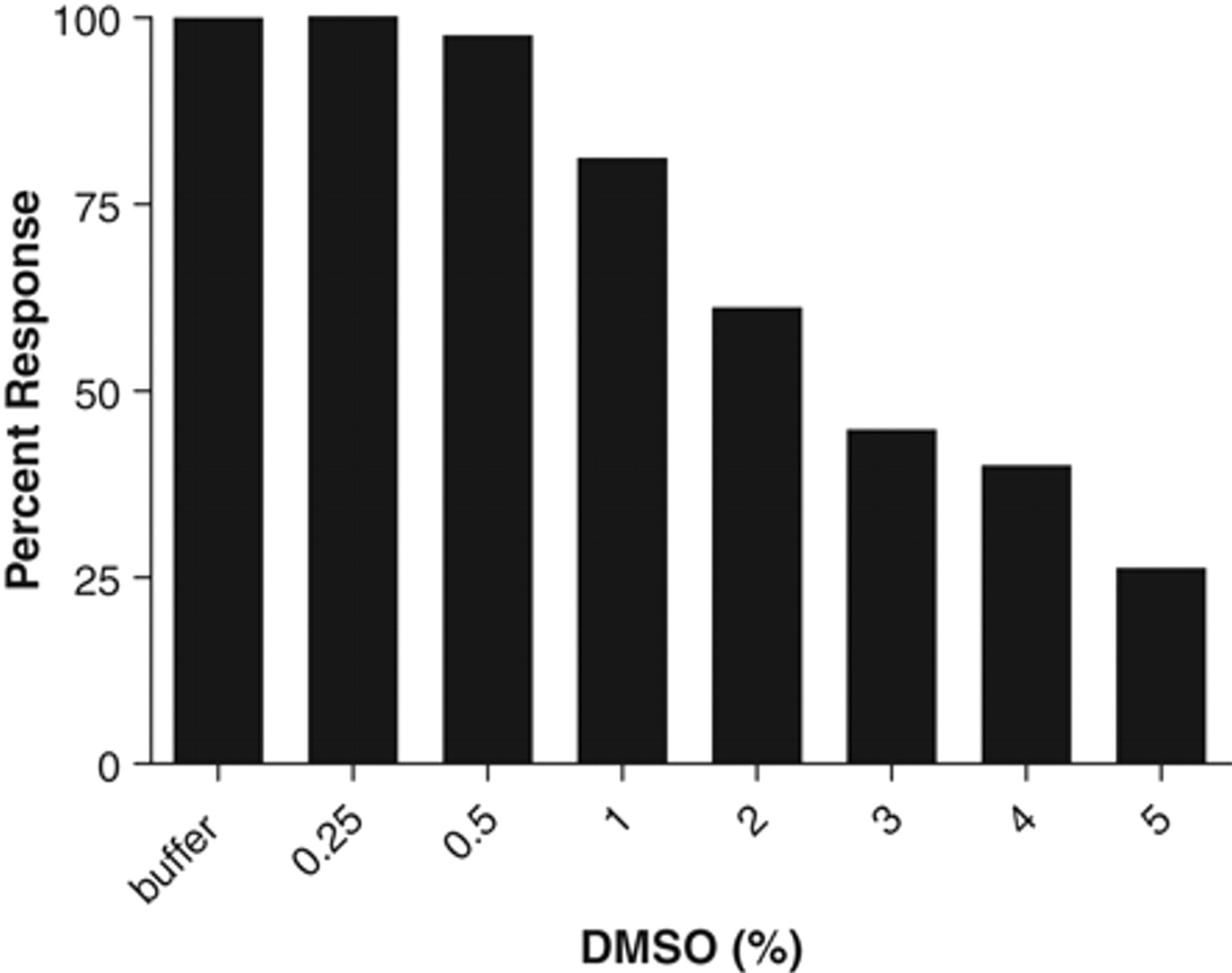
Effects of DMSO concentrations on Tl+ influx signal. DMSO was tested from 0.25% to 5% in the final assay volume, and the effects were expressed as a percentage of the response with buffer alone, which was set as 100%.
HTS is often performed using a mixture of compounds in each well of a multiwell plate, and in this situation, it is a routine practice to assess whether the positive control can be identified in the mixed-compound format, thus providing a measure of the sensitivity of the assay. For this purpose, 10 µM of staurosporine was spiked into 2 wells of an assay plate with mixtures of 10 compounds at 2.5 µM. Staurosporine-stimulated Tl+ influx was clearly observed in the 2 wells where staurosporine was deposited ( Fig. 7 ). These data suggest that the Tl+ influx assay can be used in a mixed-compound format for HTS.
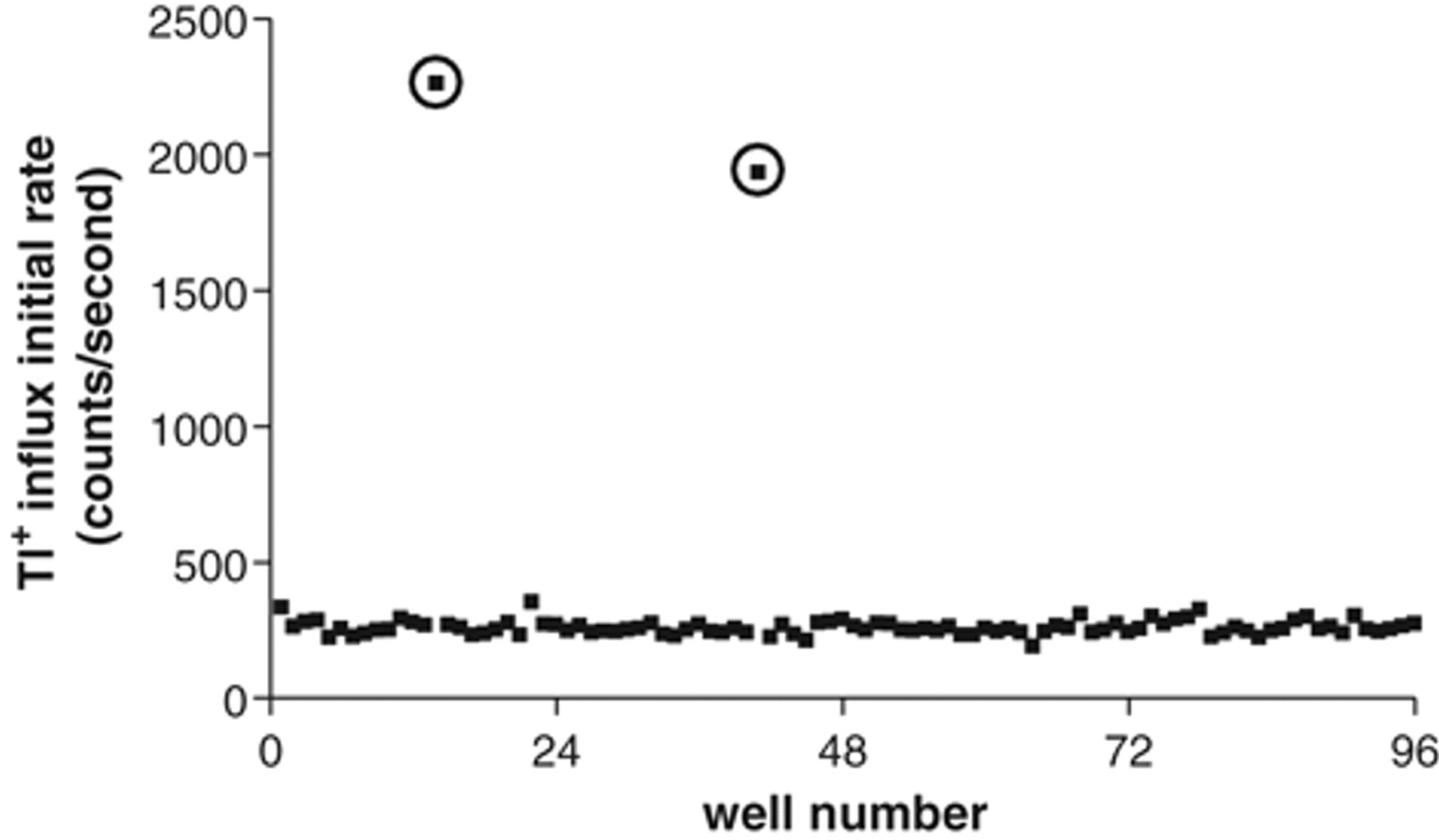
Scatter plot of Tl+ influx signals from a test plate in which 10 µM staurosporine was spiked into 2 wells of 10 compound mixtures at 2.5 µM. The signals from the 2 wells where staurosporine was deposited are circled.
Discussion
Neuronal-specific K-Cl cotransporter KCC2 represents a potentially important drug target for CNS disorders such as epilepsy and neuropathic pain. Here we describe a Tl+ transport assay that can be employed to screen for positive modulators of KCC2 in high-throughput format. This assay is feasible owing to the fact that KCC2 is not highly selective for one particular cation and is able to transport Tl+ ions, for which a fluorescence-based ion flux indicator is available. In a similar Tl+ flux assay developed to measure potassium channel activity, 19 due to the poor solubility of Tl+ in Cl−-containing buffer, a Cl−-free buffer was used. However, we could not perform the KCC2 assay in Cl−-free buffer because the transport of Tl+ by KCC2 requires coupled transport of Cl− in the same direction. Fortunately, we determined that KCC2 has a high affinity for external Tl+, and a robust FLIPR signal can be initiated with just 1 mM Tl+, at which concentration Tl+ is still soluble in assay buffer with physiological Cl− concentration. The affinity of KCC2 for Tl+ (Km = 249 µM) is greater than the reported affinity for K+/Rb+ (Km ~5-9 mM). 11,21 This may explain our observation that Tl+ influx driven by KCC2 is not sensitive to external K+ up to 10 mM but can be competed by K+ at higher concentrations. The significantly higher affinity for external K+/Tl+ versus Cl− ions observed for KCC2-mediated transport may be a reflection of its possible dual role in the buffering of external K+ ion concentration in neurons, 11 in addition to its role in Cl− homeostasis necessary for fast inhibitory neurotransmission and the higher physiological external Cl− concentration versus that of K+ ion.
Several observations led us to believe that the Tl+ influx is mediated by KCC2. First, substantial Tl+ influx was observed in KCC2-expressing cells but not in mock-transfected cells, and the influx signals correlate with the amounts of KCC2 expressed. Second, the Tl+ influx exhibited dependency on external Cl−, a characteristic of KCC2 as a K-Cl cotransporter. Third, stimulation by NEM and inhibition by DIOA are two major characteristics of KCC transporters. 1,24 Consistent with this, we observed stimulation of Tl+ influx signal by known positive KCC modulators NEM and staurosporine in KCC2-expressing cells and inhibition of the signal by DIOA and furosemide with expected potencies.
One layer of complexity is that HEK293 cells express an NKCC-like activity endogenously, and this activity, if not blocked, would contribute to the Tl+ influx signal. 20 Thus, we performed the Tl+ influx assay in the presence of 10 µM bumetanide to abolish any NKCC activity but leave KCC2 activity intact, according to the differential potency reported for bumetanide at these 2 cotransporters. 11,20-22 Under such conditions, we observed substantially higher Tl+ influx signal in HEK293: hKCC2 cells compared to that in HEK293 cells. In addition, the signal can be inhibited by DIOA with an IC50 value characteristic of KCC2 but not NKCC. 26 Therefore, the Tl+ influx observed in our assay is due to KCC2 activity. While our manuscript was in preparation, Delpire et al. 22 published a similar Tl+ influx assay for screening of KCC2 inhibitors. However, they did not block the NKCC-like activity that they detected as expressed endogenously in their HEK293-based KCC2 cell line and reported a high proportion of false positives on screening for KCC2 modulators.
The Tl+ influx assay offers several significant advantages over other functional assays to measure KCC2 activity. Compared to an anion reversal potential assay, 14 this assay offers much higher throughput and measures ion flux directly. Compared to radio- active and nonradioactive Rb+ flux assays, 11,27 the Tl+ influx assay offers high temporal resolution with measurement of real-time changes in ion flux profile within the initial seconds, where ion transport is occurring at a linear rate, rather than minutes, as in the Rb+ flux assays. Importantly, as a continuous assay, the Tl+ flux assay is easier to perform and inherently offers improved sensitivity and accuracy over endpoint assay. Compared to a NH4 + uptake assay measured with a pH-sensitive fluorescent dye, 15 the Tl+ influx assay is a more direct and simple assay with better resolution and sensitivity. Finally, our Tl+ influx assay is distinguished from that recently published by Delpire et al. 22 in that modulators more specific to KCC2 could be identified with fewer false positives by screening under our conditions, in which background cotransporter activity is efficiently blocked.
Conclusion
We have developed and validated a FLIPR-based Tl+ transport assay in which KCC2 activity is directly assessed, in a continuous assay, by the initial rate of KCC2-mediated Tl+ influx. We further provide evidence that the assay is robust, reproducible, and suitable for the identification of positive KCC2 modulators in high-throughput format. In addition to utility for KCC2, this or similar Tl+ transport assays could potentially be used to measure the activities and to identify modulators of other K-Cl cotransporters.
Footnotes
Acknowledgements
The authors thank Drs. Murali Gopalakrishnan and Michael Jarvis for their helpful suggestions during the course of this research.