
Abstract
Introduction
ADHD commonly persists into adulthood (Barkley, Fischer, Smallish, & Fletcher, 2002; Mannuzza, Klein, Bessler, Malloy, & LaPadula, 1998; Weiss, Hechtman, Milroy, & Perlman, 1985). Adult ADHD is associated with negative effects on daily functioning and goal attainment, including impaired educational, occupational, and social functioning (Biederman et al., 2006; de Graaf et al., 2008). In adults, psychostimulants are the most commonly prescribed ADHD medication (Castle, Aubert, Verbrugge, Khalid, & Epstein, 2007), and psychostimulants approved for use in adults in the United States are sustained-release formulations. Overall, currently approved medications exhibit durations of effect of 8 to 14 hr (Adler et al., 2017; Thomas, Rostain, & Prevatt, 2013). However, some adults with ADHD may require a medication with a more prolonged duration of effect.
Achieving symptom control into the evening hours for adults with ADHD may require a dose-augmentation strategy (e.g., an extended-release formulation in the morning followed by an immediate-release formulation later in the day) (Adler, Reingold, Morrill, & Wilens, 2006). However, medication adherence may be limited by the core ADHD symptoms (forgetfulness, failure to follow instructions, and disorganization) (American Psychiatric Association, 2013) and by embarrassment about taking medication during the workday (Swanson, 2003). Although adherence to psychostimulants, and to medication in general, may be inversely proportional to the frequency of required dosing (Dodson, 2005; Eisen, Miller, Woodward, Spitznagel, & Przybeck, 1990), support for this concept is equivocal. One study reported improved adherence after participants were switched from 3 times daily, immediate-release methylphenidate (MPH) to once-daily, extended-release MPH (Ramos-Quiroga et al., 2008). In another study, adherence did not significantly differ between extended-release mixed amphetamine salts (MAS XR) and immediate-release MAS (MAS IR) in adults based on pill count and self-report. However, when adherence was assessed using a computerized monitoring device, MAS XR was associated with significantly greater adherence compared with MAS IR (Adler et al., 2011).
Triple-bead MAS, also known as SHP465, is a once-daily, triple-bead, sustained-release, single-entity MAS product for oral administration. It is comprised of sulfate salts of d-amphetamine and amphetamine, with d-amphetamine saccharate and amphetamine aspartate monohydrate. The capsule contains three types of drug-releasing beads, which provide for immediate release, pulsatile delayed release, and sustained-release of the MAS. In a Phase 1 study, the pharmacokinetic profile of 37.5 mg triple-bead MAS was similar to that of a morning dose of 25 mg MAS XR supplemented 8 hr later by 12.5 mg MAS IR (Ermer et al., 2007). In a 7-week, Phase 3, dose-optimization study in adults with ADHD, triple-bead MAS (12.5-75 mg) produced significantly greater reductions in ADHD Rating Scale IV (ADHD-RS-IV) total score from baseline than placebo—least squares (LS) mean (95% confidence interval [CI] treatment difference at end of study [EOS]): −8.1 [−10.8, −5.4]) (Spencer, Adler, Weisler, & Youcha, 2008). Mean ± SD increases in systolic blood pressure (SBP), diastolic blood pressure (DBP), and pulse at EOS with triple-bead MAS were 1.3 ± 9.3 mmHg, 1.8 ± 6.7 mmHg, and 4.7 ± 11.1 bpm, respectively (Spencer et al., 2008). Insomnia (29.2%) was the most commonly reported treatment-emergent adverse event (TEAE) with triple-bead MAS (Spencer et al., 2008).
The primary objective of this study was to evaluate the efficacy of fixed doses of triple-bead MAS (25, 50, or 75 mg; individually and combined across doses) versus placebo. The primary efficacy assessment was the treatment difference for the change from baseline in ADHD-RS-IV total score at EOS versus placebo. The key secondary objective was to assess the efficacy dose response of triple-bead MAS based on the change from baseline at EOS in ADHD-RS-IV total score. Safety and tolerability assessments of triple-bead MAS were included as secondary endpoints.
Materials and Method
Study Design
This randomized, placebo-controlled, double-blind, forced-dose study (ClinicalTrials.gov registry number: NCT00152022) was conducted at 48 U.S. sites between April 25, 2005, and November 4, 2005. It included a 2-week screening phase, a 1- to 4-week washout phase, a 6-week forced-dose double-blind treatment phase, and a 30-day (±5 days) follow-up period. The protocol was approved by a central institutional review board (IRB; 43 of 48 sites) or by local IRBs (5 of 48 sites). The study was managed by Quintiles Inc. (San Diego, CA), which was responsible for submitting all study-related information to the central IRB; sites using local IRBs submitted all study-related documents directly to the central IRB.
The study was conducted in accordance with the principles of the World Medical Association Declaration of Helsinki (World Medical Association, 2001), including amendments of the 29th, 35th, 41st, and 48th World Medical Assemblies. Participants received a study description and provided written informed consent before participating; all informed consent documents were written in accordance with Good Clinical Practice and guidelines established by the 1996 Health Insurance Portability and Accountability Act.
Participants
Adults (men or nonpregnant, nonlactating women aged 18-55 years) meeting Diagnostic and Statistical Manual of Mental Disorders (4th ed., text rev.; DSM-IV-TR; American Psychiatric Association, 2000) criteria for a primary ADHD diagnosis established using the Adult ADHD Clinical Diagnostic Scale Version 1.2 and having baseline ADHD-RS-IV (Adler, Spencer, et al., 2009) total scores ≥32 were eligible. All eligible participants had satisfactory medical assessments, with no clinically significant or relevant abnormalities.
Key exclusion criteria included current comorbid psychiatric disorders (defined by the Structured Clinical Interview for the DSM-IV-TR [SCID] Axis I Disorders and controlled with prohibited medications or uncontrolled and associated with significant symptoms); any conditions/symptoms that could confound clinical assessments at screening; chronic or acute illnesses or unstable medical conditions that could confound safety assessments, lead to increased risk, or make it difficult to comply with the protocol; a history of seizures (excluding infantile febrile seizures), any tic disorder, or a current diagnosis and/or family history of Tourette disorder; known cardiac abnormalities or conditions affecting cardiac performance, a history of hypertension, or sitting SBP >139 mmHg or DBP >89 mmHg; a clinically significant electrocardiogram (ECG) or laboratory abnormality at baseline or screening; the use of a psychoactive prescription medication or over-the-counter medication requiring more than a 28-day washout period (excluding hormonal contraceptives); participation in a clinical study within 30 days of screening; a drug dependence or substance abuse disorder (excluding SCID-defined nicotine dependence) within 6 months before screening or a positive urine drug result at screening or baseline (excluding current psychostimulant medications); and a documented allergy, intolerance, or history of nonresponse to MPH or amphetamine.
Treatment
After screening and washout, participants were randomized (1:1:1:1) to 6 weeks of forced-dose triple-bead MAS (25, 50, or 75 mg) or matching placebo (Figure 1A). The triple-bead MAS dosage during the first double-blind treatment week was 25 mg for all participants. Participants randomized to 25 mg triple-bead MAS remained on this dosage for the duration of treatment. The dosage for participants randomized to 50 or 75 mg triple-bead MAS was force titrated over 2 to 3 weeks (50-mg group: 37.5 mg during Week 2, 50 mg during Weeks 3-6; 75-mg group: 37.5 mg during Week 2, 50 mg during Week 3; 75 mg during Weeks 4-6). Modification of the titration schedules was not allowed. During the follow-up period, telephone contact was initiated to assess ongoing or new adverse events (AEs). If AEs were not acceptably resolved, follow-up continued until concerns resolved or the participant was lost to follow-up.
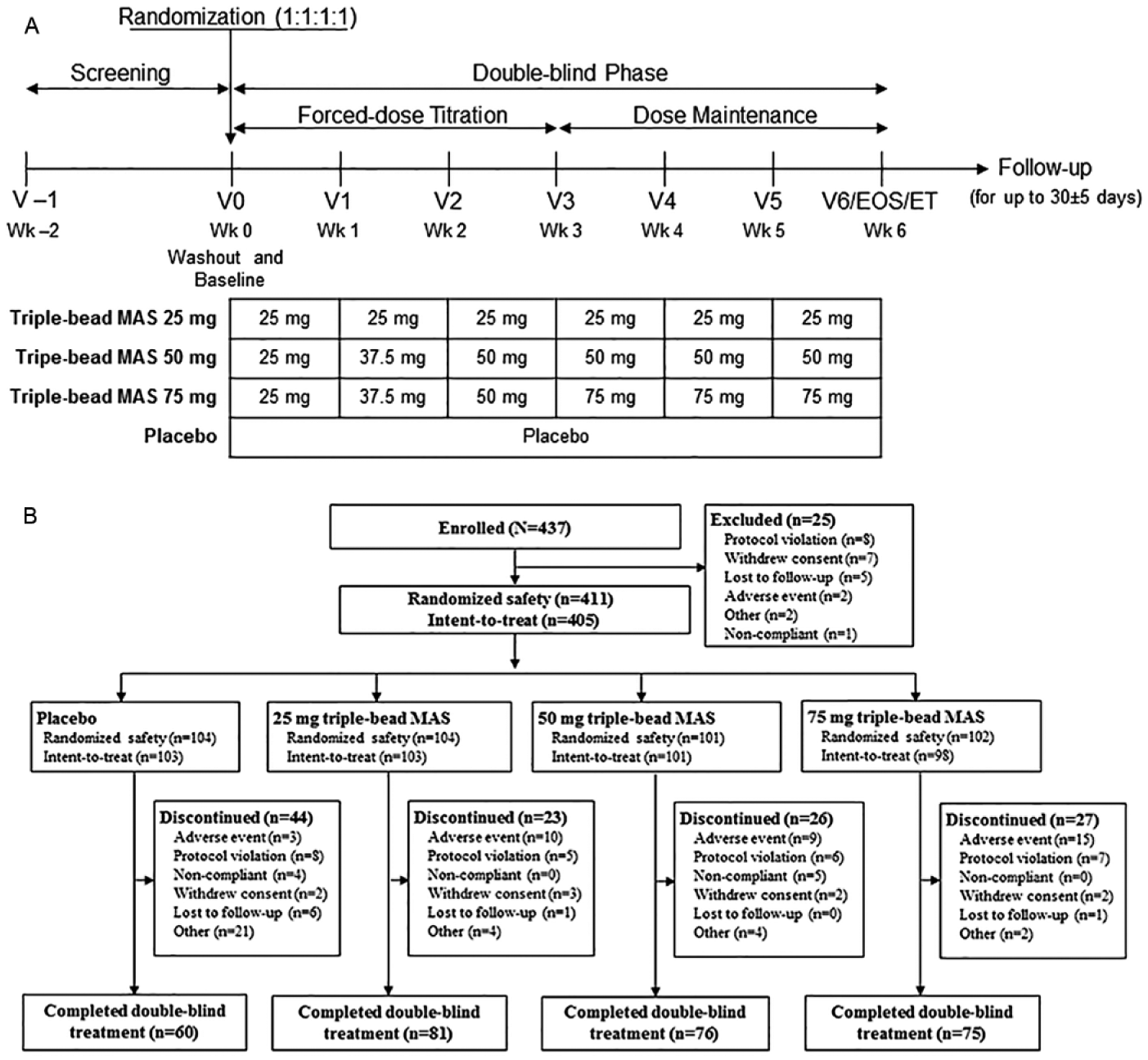
Study design (A) and participant disposition (B).
Treatment was determined by a randomization schedule prepared by Quintiles Inc. Computer-generated two-part labels were used for blinding, and all treatments were identical in appearance. The blind was inadvertently broken for four participants during the study (three participants had urine drug screens required by their employers or housing development; one participant had a urine sample erroneously processed as a baseline urine drug screen sample).
Efficacy Endpoints
The primary efficacy endpoint was ADHD-RS-IV total score change from baseline to EOS, with EOS for the primary efficacy endpoint defined as the average of treatment Weeks 4 to 6 or to the last postrandomization treatment assessment for which a valid assessment was obtained. The ADHD-RS-IV was assessed at screening, baseline (Visit 0), and Weeks 1 (Visit 1) through 6 (Visit 6/EOS). The ADHD-RS-IV is an 18-item, clinician-administered scale (DuPaul, Power, Anastopoulos, & Reid, 1998); severity for each item is scored from 0 (none) to 3 (severe), and total score ranges from 0 to 54. A prespecified key secondary endpoint was the assessment of the dose–response relationship for triple-bead MAS on ADHD-RS-IV total score at EOS.
Although the ADHD-RS-IV was developed for children, with training and the use of prompts, clinicians can use the ADHD-RS-IV to assess the adult ADHD symptoms and examine the extent of those symptoms (Adler & Cohen, 2004; Murphy & Adler, 2004). The use of the ADHD-RS-IV with adult prompts has been validated (Adler, Spencer, et al., 2009) and has been shown to be effective in assessing treatment effects in adults with ADHD (Adler et al., 2008; Spencer et al., 2007; Weisler et al., 2009; Wilens et al., 2005). In this study, training on the use of the ADHD-RS-IV with adult prompts was conducted using standardized methods (Adler et al., 2005) in a manner consistent with a previous study of triple-bead MAS (Spencer et al., 2008).
Other secondary efficacy endpoints included the Clinical Global Impressions–Improvement (CGI-I), the Brown Attention Deficit Disorder Scales (BADDS), and the ADHD Impact Module–Adult (AIM-A). The CGI-I measures improvement in global symptom severity (Guy, 1976) on a 7-point scale (1 = very much improved to 7 = very much worse; 0 indicates “not assessed”), with improvement measured against baseline CGI–Severity scores. Scores on the CGI-I were assessed at Weeks 1 through 6/early termination (ET; the last nonmissing postbaseline assessment). The results of the BADDS and the AIM-A data from this study have previously been reported (Brown & Landgraf, 2010); none of the data included in the current report have previously been published.
Safety and Tolerability Endpoints
Assessment of AEs was initiated at the time of informed consent and took place at all subsequent study visits and at follow-up. Vital signs and 12-lead ECGs were assessed at screening, baseline, and Weeks 1 through 6/ET; midweek visits (4 ± 1 days after the Week 3 visit) were required for all treatment groups to repeat vital sign assessments when titration was complete. Clinical laboratory evaluations were assessed at screening, baseline (only urine drug test screen and urine pregnancy test), and Week 6; abbreviated baseline physical examinations and clinical laboratory tests were required if more than 30 days had elapsed since screening.
Sleep quality was assessed at baseline and Weeks 1 through 6 using a modified 18-item Pittsburgh Sleep Quality Index (PSQI). Individual PSQI items can be used to generate a global score and seven component scores (Backhaus, Junghanns, Broocks, Riemann, & Hohagen, 2002; Buysse, Reynolds, Monk, Berman, & Kupfer, 1989); PSQI global scores range from 0 to 21 (higher scores reflect worse sleep quality).
Data Presentation and Statistics
Sample size was estimated based on an unpublished effect size (ES) analysis of MAS XR data from a previously published study (Weisler et al., 2006), where an ES of 0.424 (based on a mean difference of 5.3 and standard deviation of 12.5) was observed between 60 mg MAS XR and 20 mg MAS XR in individuals with baseline ADHD-RS-IV total scores ≥32. Based on this unpublished analysis, the ES for the treatment differences between 75 mg triple-bead MAS and placebo and between 75 mg triple-bead MAS and 25 mg triple-bead MAS was assumed to be 0.424 for the sample size estimate. Assuming a dropout rate of 15%, it was estimated that 103 participants per treatment would provide 80% power.
Statistical assessment of the primary and secondary efficacy endpoints was conducted in the intent-to-treat population (all randomized participants taking ≥1 study drug dose and having ≥1 postbaseline primary efficacy assessment and a baseline assessment). Statistical significance was set at p < .05. Because the study was not powered for the secondary endpoints, only nominal unadjusted p values are reported for these endpoints; these values are included for descriptive purposes only.
Change in ADHD-RS-IV total score from baseline to EOS (primary endpoint) was assessed using a one-way ANCOVA, with treatment as a factor and baseline score as a covariate. A hierarchical testing procedure was used for assessment of the primary efficacy endpoint, with testing conducted in the following order: placebo versus the combined triple-bead MAS treatment groups (total triple-bead MAS), placebo versus 75 mg triple-bead MAS, placebo versus 50 mg triple-bead MAS, and placebo versus 25 mg triple-bead MAS. A later test could only attain significance if all earlier tests were significant. ES was calculated post hoc as the LS mean difference divided by the square root of mean squared error.
The triple-bead MAS dose–response relationship was assessed using contrasts that compared LS mean treatment differences for the ADHD-RS-IV total score change from baseline to EOS between individual triple-bead MAS dosages. Scores on the CGI-I were dichotomized as improved (scores of 1 or 2 [very much/much improved]) versus not improved (scores of 3-7 [minimally improved through very much worse]) and were analyzed using pairwise chi-square tests. Changes from baseline to EOS on the ADHD-RS-IV subscales were assessed using the one-way ANCOVA model used for the primary endpoint.
Safety and tolerability endpoints are presented for the randomized safety population (all randomized participants who took ≥1 study drug dose, including those who were randomized and dispensed study medication but immediately lost to follow-up) using descriptive statistics.
Results
Participant Disposition and Demographics
Participant disposition and demographic and clinical characteristics are summarized in Figure 1B and Table 1, respectively. Most participants were White men; mean age was 37.1 years across all groups. The mean duration of ADHD diagnosis ranged from 4.3 (25 mg triple-bead MAS) to 5.2 (placebo and 50 mg triple-bead MAS) years. Mean ADHD-RS-IV total scores at baseline ranged from 39.9 (25 mg triple-bead MAS) to 40.9 (50 mg triple-bead MAS). Most study participants were diagnosed as having the combined ADHD subtype; the distribution of ADHD subtypes across treatment groups was generally similar.
Baseline Demographic and Clinical Characteristics, Randomized Safety Population.
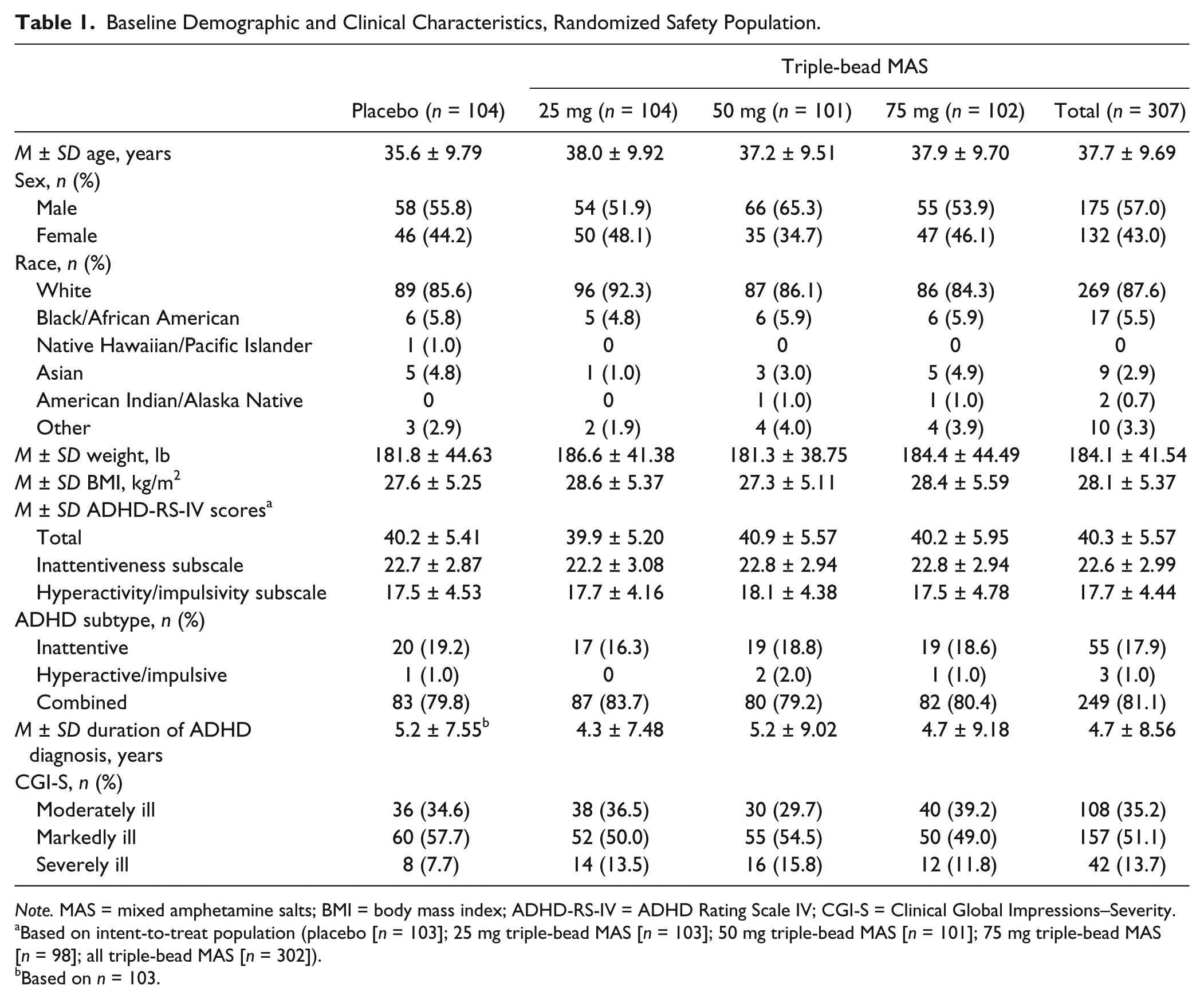
Note. MAS = mixed amphetamine salts; BMI = body mass index; ADHD-RS-IV = ADHD Rating Scale IV; CGI-S = Clinical Global Impressions–Severity.
Based on intent-to-treat population (placebo [n = 103]; 25 mg triple-bead MAS [n = 103]; 50 mg triple-bead MAS [n = 101]; 75 mg triple-bead MAS [n = 98]; all triple-bead MAS [n = 302]).
Based on n = 103.
Prior Medications
In the randomized safety population, 16.3% (67/411) of participants reported using ADHD medications within 30 days of screening (placebo, 14.4% [15/104]; 25 mg triple-bead MAS, 12.5% [13/104]; 50 mg triple-bead MAS, 16.8% [17/101]; 75 mg triple-bead MAS, 21.6% [22/102]); ADHD medications used by ≥2% of participants were MAS XR (9.7% [40/411]) and MAS IR (2.2% [9/411]).
Exposure
Mean ± SD triple-bead MAS daily dosage (all doses combined) during the entire double-blind treatment phase was 38.7 ± 12.71 mg. For the individual triple-bead MAS groups, mean ± SD dosages over the course of treatment were 25.0 ± 0.00 mg (25 mg), 41.2 ± 5.46 mg (50 mg), and 50.3 ± 11.15 mg (75 mg). Mean ± SD weeks of exposure to triple-bead MAS was 5.21 ± 1.780 (all doses combined). Mean ± SD adherence (number of capsules taken divided by the number of capsules that should have been taken over the treatment period) was 97.2% ± 10.52% with placebo and 97.1% ± 13.03% with triple-bead MAS (range: 94.9% ± 19.45% with 75 mg triple-bead MAS to 98.6% ± 3.86% with 25 mg triple-bead MAS).
Efficacy
Primary endpoint: Change in ADHD-RS-IV total score
Reductions in ADHD-RS-IV total score were observed in all treatment groups (Table 2, Figure 2A). At EOS, the LS mean treatment differences significantly favored all triple-bead MAS doses (all doses combined and each individual dose) over placebo when assessed by randomized treatment group (Table 2) and by actual dose (Figure 2B). Individual dose ESs favoring triple-bead MAS ranged from 0.85 to 0.96 (Table 2). Nominally significant LS mean (95% CI) treatment differences (triple-bead MAS—placebo) were observed as early as Week 1 (25-mg dose, −5.8 [−8.0, −3.5]; p < .0001).
Summary of Efficacy Endpoints by Randomized Dose, Intent-to-Treat Population.
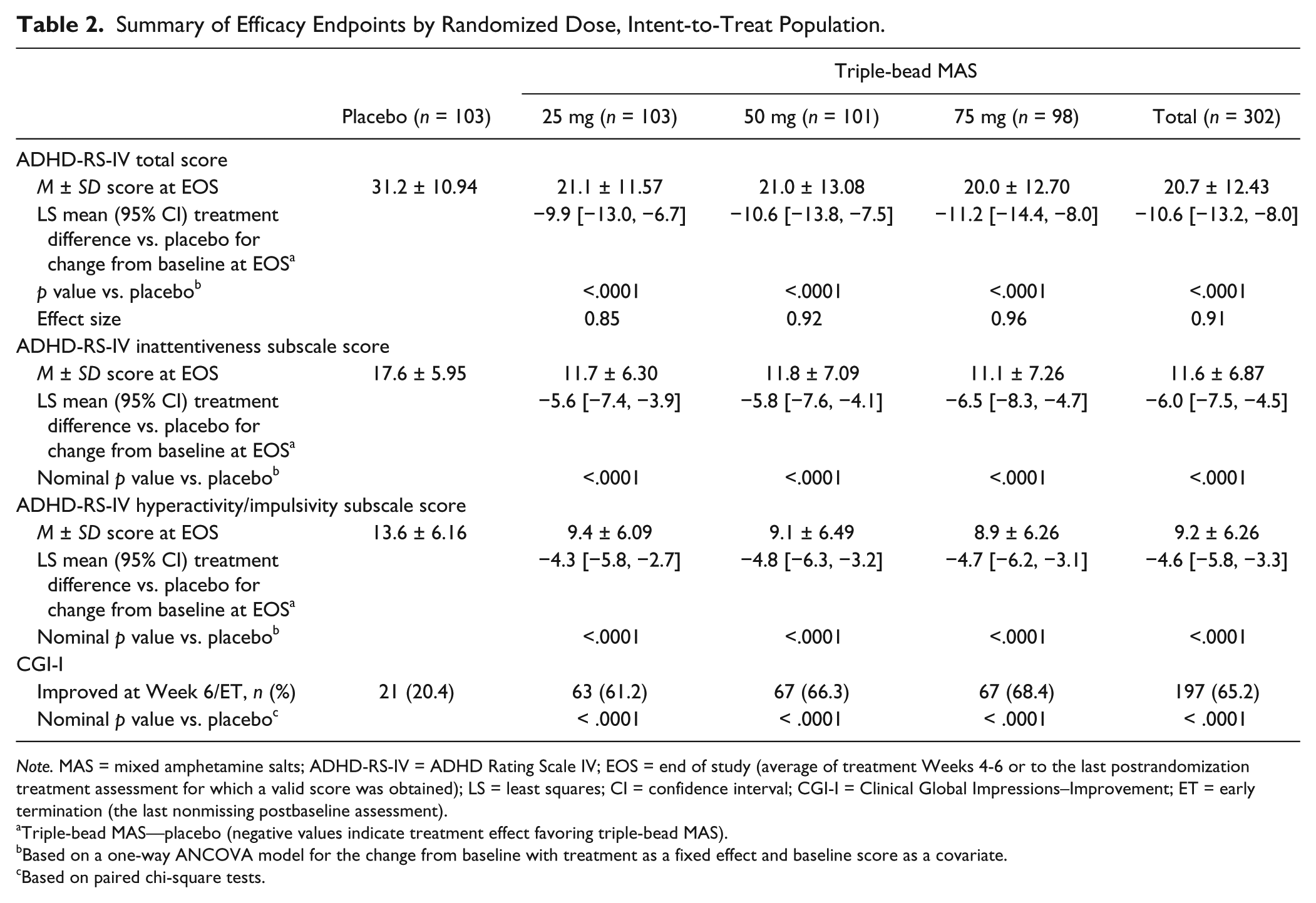
Note. MAS = mixed amphetamine salts; ADHD-RS-IV = ADHD Rating Scale IV; EOS = end of study (average of treatment Weeks 4-6 or to the last postrandomization treatment assessment for which a valid score was obtained); LS = least squares; CI = confidence interval; CGI-I = Clinical Global Impressions–Improvement; ET = early termination (the last nonmissing postbaseline assessment).
Triple-bead MAS—placebo (negative values indicate treatment effect favoring triple-bead MAS).
Based on a one-way ANCOVA model for the change from baseline with treatment as a fixed effect and baseline score as a covariate.
Based on paired chi-square tests.
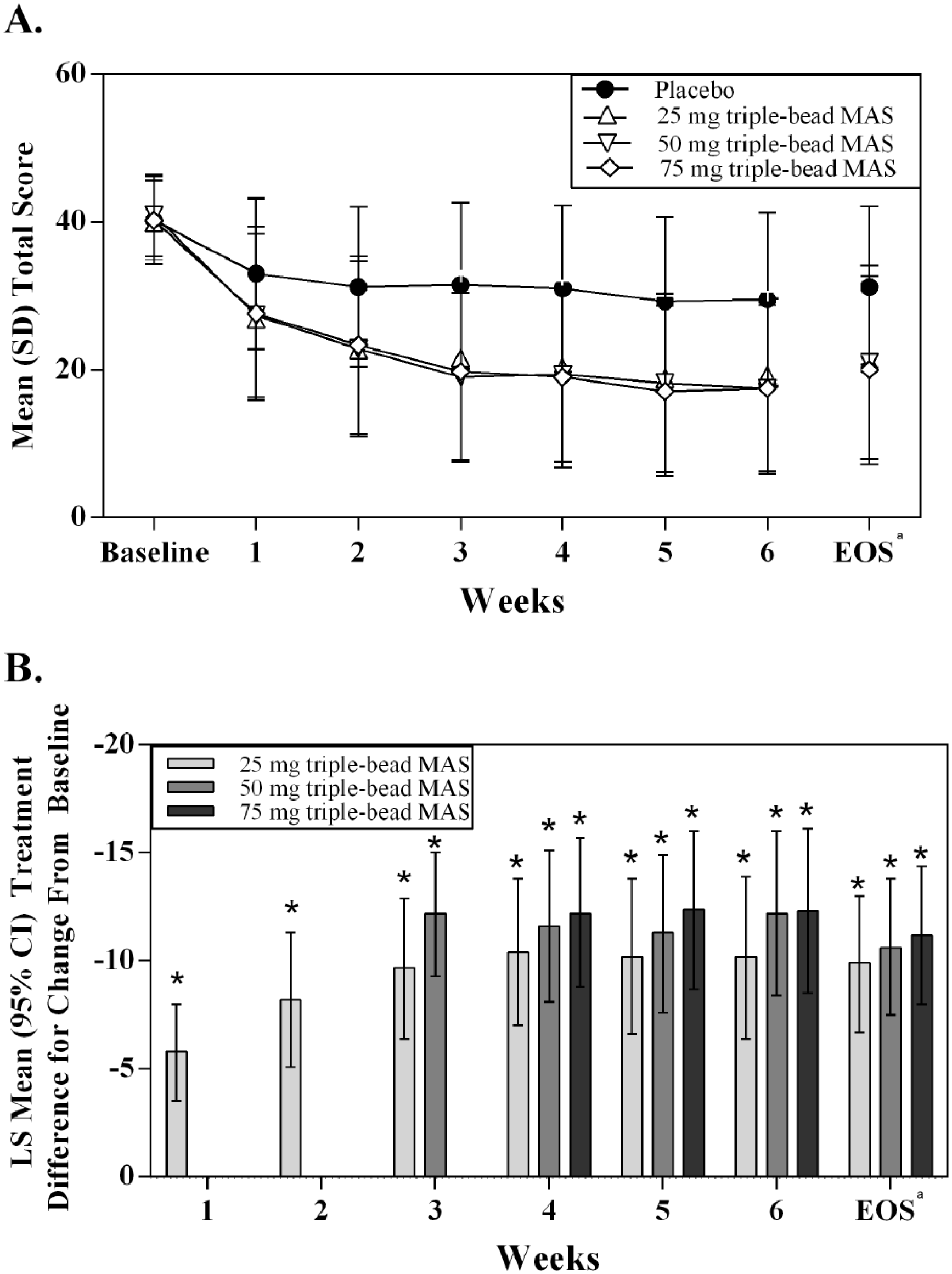
ADHD Rating Scale IV total scores (A) and treatment differences versus placebo based on actual dose (B).
Key secondary endpoint: Triple-bead MAS dose response
The LS mean (95% CI) treatment differences for change from baseline at EOS between triple-bead MAS randomized treatment groups did not differ statistically (25 vs. 50 mg: −0.8 [−4.0, 2.4]; 25 vs. 75 mg: −1.3 [−4.5, 1.9]; 50 vs. 75 mg: −0.5 [−3.8, 2.7]). Similarly, LS mean (95% CI) treatment differences for change from baseline at Week 6 between triple-bead MAS dosages based on the actual dose did not differ statistically (25 vs. 50 mg: −2.0 [−5.5, 1.5]; 25 vs. 75 mg: −2.1 [−5.6, 1.4]; 50 vs. 75 mg: −0.1 [−3.7, 3.5]).
Other secondary endpoints
On both ADHD-RS-IV subscales, LS mean treatment differences for change from baseline at EOS nominally favored triple-bead MAS (for all doses combined and for each individual dose) over placebo (Table 2). On the dichotomized CGI-I, nominally greater percentages of participants were categorized as improved at Week 6/ET with triple-bead MAS than with placebo (Table 2; all nominal p < .0001).
Safety and Tolerability Endpoints
A majority of participants exhibited TEAEs, most of which were of mild or moderate severity (Table 3). A dose–response relationship was observed for severe TEAEs, with increasing triple-bead MAS doses associated with higher percentages of severe TEAEs. Severe TEAEs reported by ≥2 participants were insomnia (n = 11; one [1.0%] with placebo, four [3.8%] with 25 mg triple-bead MAS, three [3.0%] with 50 mg triple-bead MAS, and three [2.9%] with 75 mg triple-bead MAS), headache and anxiety (n = 3; one [1.0%] each with placebo, 50 mg triple-bead MAS, and 75 mg triple-bead MAS), and fatigue (n = 2; 1 [1.0%] each with 25 and 75 mg triple-bead MAS). The percentage of severe insomnia-related TEAEs was greater with triple-bead MAS (initial insomnia: 0.3% [1/307]; insomnia: 3.3% [10/307]) than with placebo (insomnia: 1% [1/104]), but no dose–response relationship was observed for triple-bead MAS. The majority of severe TEAEs resolved, with the exception of five instances in four participants (25 mg triple-bead MAS: insomnia; 50 mg triple-bead MAS (onset at 25 mg): sleep disorder [verbatim of poor sleep]; 50 mg triple-bead MAS: insomnia; 75 mg triple-bead MAS: dry mouth and dysgeusia). One serious TEAE (a puncture wound in the hand) was reported by a participant randomized to 75 mg triple-bead MAS and was deemed unrelated to study medication by the investigator. There were no deaths during the trial.
Summary of TEAEs, Randomized Safety Population.
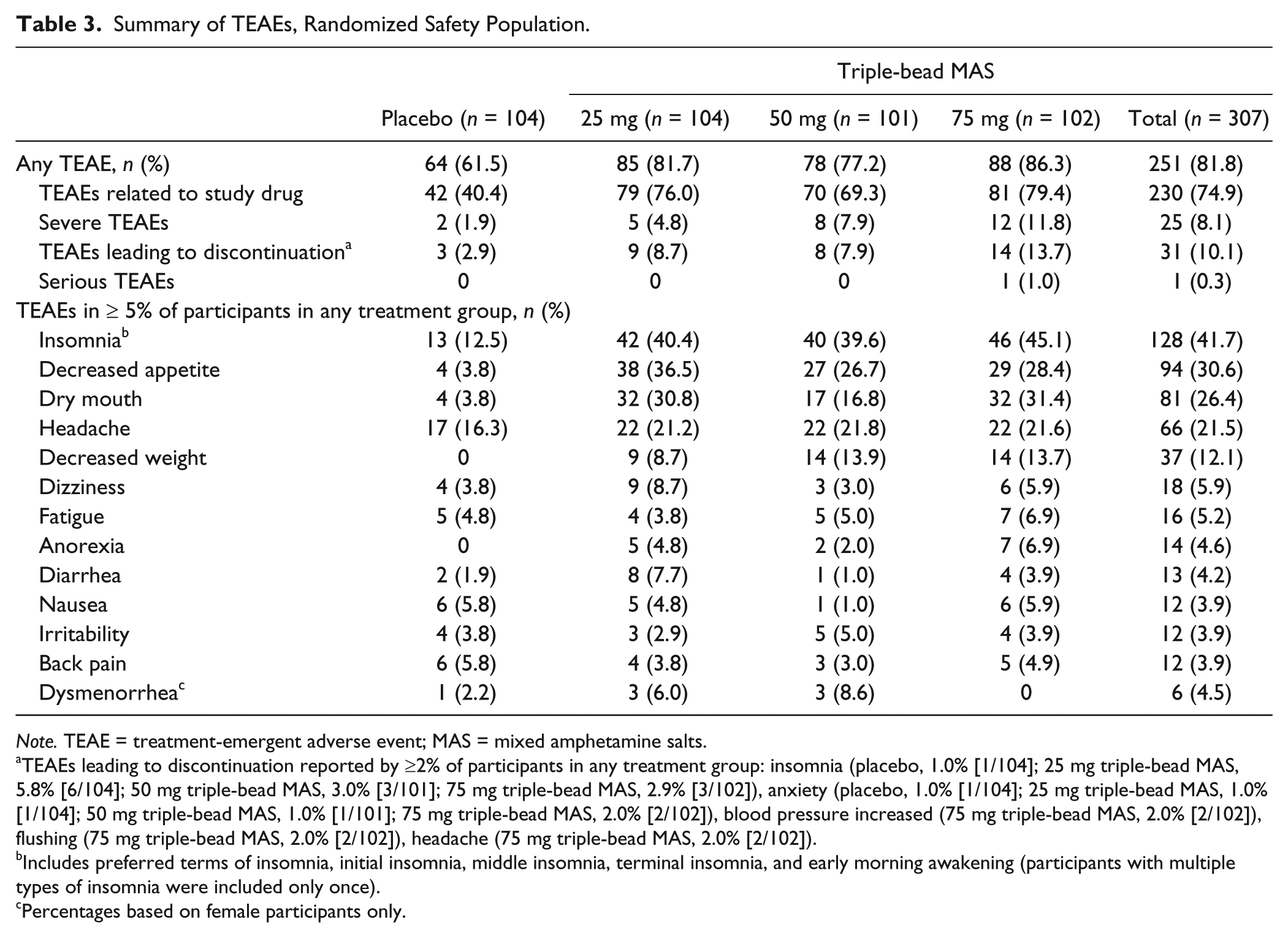
Note. TEAE = treatment-emergent adverse event; MAS = mixed amphetamine salts.
TEAEs leading to discontinuation reported by ≥2% of participants in any treatment group: insomnia (placebo, 1.0% [1/104]; 25 mg triple-bead MAS, 5.8% [6/104]; 50 mg triple-bead MAS, 3.0% [3/101]; 75 mg triple-bead MAS, 2.9% [3/102]), anxiety (placebo, 1.0% [1/104]; 25 mg triple-bead MAS, 1.0% [1/104]; 50 mg triple-bead MAS, 1.0% [1/101]; 75 mg triple-bead MAS, 2.0% [2/102]), blood pressure increased (75 mg triple-bead MAS, 2.0% [2/102]), flushing (75 mg triple-bead MAS, 2.0% [2/102]), headache (75 mg triple-bead MAS, 2.0% [2/102]).
Includes preferred terms of insomnia, initial insomnia, middle insomnia, terminal insomnia, and early morning awakening (participants with multiple types of insomnia were included only once).
Percentages based on female participants only.
The TEAEs leading to discontinuation reported by ≥2% of participants in any treatment group (see footnote ‘a’ to Table 3) included insomnia, anxiety, increased blood pressure, flushing, and headache. The TEAEs reported by ≥5% of participants in any triple-bead MAS treatment group at twice the frequency of placebo were insomnia, decreased appetite, dry mouth, decreased weight, dizziness, dysmenorrhea, diarrhea, and anorexia. Of these TEAEs, only decreased weight showed an apparent dose response, with higher frequencies reported with 50 and 75 mg triple-bead MAS than with 25 mg triple-bead MAS.
Decreases in mean pulse, SBP, and DBP were observed with placebo at Week 6/ET when assessed by randomized treatment group (Table 4) and by actual dose (Supplemental Table). Dose-dependent increases in pulse were observed with increasing triple-bead MAS doses; both SBP and DBP decreased with 25 and 50 mg triple-bead MAS but increased with 75 mg triple-bead MAS (Table 4).
Summary of Vital Sign and Physical Examination Changes by Randomized Dose, Randomized Safety Population.
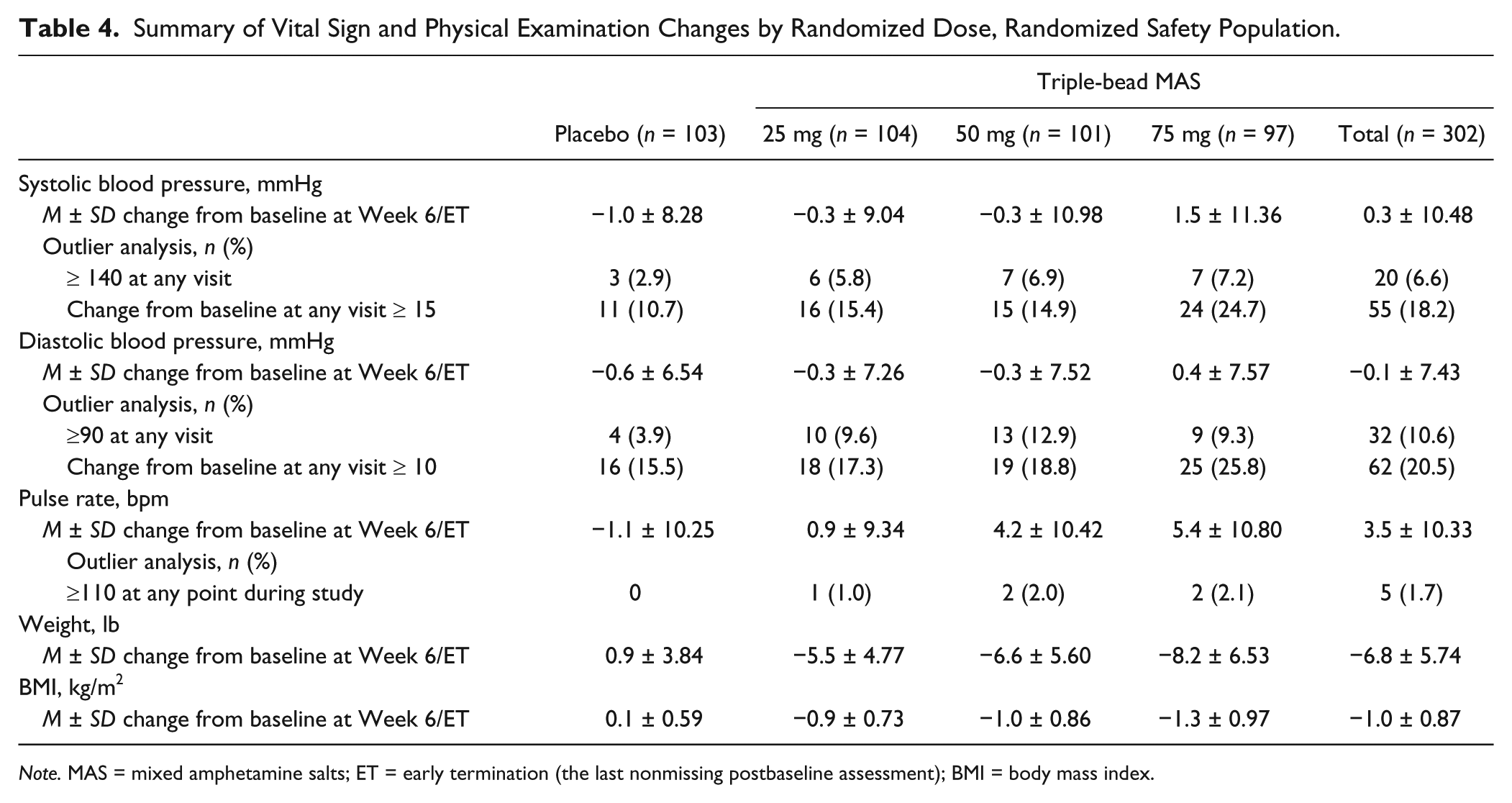
Note. MAS = mixed amphetamine salts; ET = early termination (the last nonmissing postbaseline assessment); BMI = body mass index.
Blood pressure outlier analyses by randomized treatment group (Table 4) and by actual dose (Supplemental Table) indicated that greater percentages of participants treated with triple-bead MAS exhibited SBP increases of ≥15 mmHg or SBP values ≥140 mmHg and DBP increases ≥10 mmHg or DBP values ≥90 mmHg at any visit than did those treated with placebo. Dose–response relationships were observed for DBP increase from baseline of ≥10 mmHg at any visit, with increasing triple-bead MAS doses associated with higher percentages of participants exhibiting this change (Table 4).
Pulse outlier analysis by randomized treatment group (Table 4) and by actual dose (Supplemental Table) indicated that greater percentages of participants treated with triple-bead MAS exhibited pulse rates of ≥110 bpm at any visit than did those treated with placebo. In one participant randomized to 75 mg triple-bead MAS, four consecutive postrandomization treatment week observations indicated SBP readings ≥140 mmHg and DBP readings ≥90 mmHg, which led to study discontinuation. Subsequently, the participant disclosed pre-existing untreated hypertension and reported that self-obtained blood pressure assessments had returned to their usual levels.
Decreases in Fridericia-corrected QT intervals (QTcF) were observed at Week 6/ET with placebo, and increases were observed with all triple-bead MAS doses (Supplemental Table). Outlier analyses indicated that no participant had a QTcF value ≥500 ms during the study. Mean changes in heart rate observed with an ECG were generally consistent with vital sign pulse data (Supplemental Table).
Weight and body mass index increased at Week 6/ET with placebo but decreased dose dependently with triple-bead MAS (Table 4). The percentage of participants reporting weight loss of >7% from baseline to Week 6/ET (by actual dose) was also dose dependent (25 mg: 4 [3.4%]; 50 mg: 15 [16.7%]; 75 mg: 20 [24.1%]); no placebo participants reported weight loss of ≥7% from baseline (Supplemental Table). There were no clinically important changes in laboratory findings from baseline observed in any treatment group.
Mean ± SD baseline PSQI total scores were 7.6 ± 3.31 (n = 104) in the placebo group and 7.1 ± 3.29 (n = 307) in the total triple-bead MAS group (25 mg: 6.9 ± 3.37 [n = 104]; 50 mg: 7.1 ± 3.11 [n = 101]; 75 mg: 7.3 ± 3.41 [n = 102]). Mean ± SD reductions from baseline at Week 6 were noted in PSQI total scores with placebo (−1.5 ± 2.70 [n = 60]) and triple-bead MAS (total: −1.7 ± 3.72 [n = 232]; 25 mg: −1.9 ± 3.62 [n = 81]; 50 mg: −1.4 ± 3.49 [n = 76]; and 75 mg: −1.8 ± 4.06 [n = 75]).
Discussion
The primary efficacy analysis from this study indicated that all assessed triple-bead MAS doses (25, 50, and 75 mg; individually and combined across doses) resulted in significantly greater reductions in ADHD-RS-IV total score than placebo in adults with ADHD. This finding was observed by randomized treatment group and by actual dose, suggesting that each individual triple-bead MAS dose exhibited efficacy versus placebo. Symptom reductions with triple-bead MAS were observed as early as Week 1 and continued through 6 weeks of treatment. However, as has previously been observed for MAS XR (Weisler et al., 2006), the key secondary analysis indicated there were no statistically significant differences in the change from baseline ADHD-RS-IV total score at EOS between triple-bead MAS doses. Score reductions on the ADHD-RS-IV subscales followed the same pattern observed for total score, with nominally greater improvement versus placebo observed for all triple-bead MAS doses. As measured by the CGI-I, triple-bead MAS also produced nominally greater improvement in global symptom severity than placebo. The ES for treatment differences in ADHD-RS-IV total score reductions from baseline averaged across triple-bead MAS doses in this study was 0.91, which compared favorably to the ES of 0.80 for MAS XR when averaged across doses of 20, 40, and 60 mg (Weisler et al., 2006).
These efficacy findings are consistent with those of a previously published dose-optimization study in which 7 weeks of triple-bead MAS (12.5-75 mg) produced significantly greater reductions in ADHD-RS-IV total score than placebo (Spencer et al., 2008). Although the previously published study of triple-bead MAS in adults with ADHD did not report an ES for treatment differences in the change from baseline in ADHD-RS-IV total scores between placebo and triple-bead MAS (Spencer et al., 2008), the magnitude of LS mean (95% CI) treatment difference in the previously published study (−8.1 [−10.8, −5.4]) fell within the CIs for the treatment difference reported in this study (all doses combined: −10.6 [−13.2, −8.0]). However, it should be noted that the ADHD-RS-IV total score inclusion criterion in the current study (≥32) differed from that used in the previously published flexible-dose study (≥24) (Spencer et al., 2008), and it is not clear how differences in baseline ADHD-RS-IV total scores may have influenced triple-bead MAS treatment effects. Although findings for secondary endpoints should be interpreted with caution because the study was not powered for their assessment, the dose-optimization study also reported that triple-bead MAS was superior to placebo in improving global symptom severity as measured by the CGI-I (Spencer et al., 2008).
Although numerically greater reductions from baseline to EOS in ADHD-RS-IV total scores were observed with 50 and 75 mg triple-bead MAS than with 25 mg triple-bead MAS, a statistically significant dose response was not observed. This finding is consistent with a previous publication in which a dose response for efficacy was not observed for MAS XR (20, 40, and 60 mg) in adults with ADHD in a 4-week, double-blind, fixed-dose study (Weisler et al., 2006). However, a post hoc analysis of participants with baseline ADHD-RS-IV total scores ≥32 from the same MAS XR study demonstrated that treatment differences in ADHD-RS-IV total score reductions with 60 mg MAS XR were significantly greater than with 20 mg MAS XR (p = .001) and approached significance compared with 40 mg MAS XR (p = .10) (Weisler et al., 2006). Possible contributing factors to the lack of a dose response in this study may be related to the use of forced-dose design, which may not allow for the optimum balance between efficacy and tolerability to be obtained in individual participants, and to the steps used in the titration schedule.
The short-term safety and tolerability of triple-bead MAS was generally similar to previously reported findings for triple-bead MAS (Spencer et al., 2008) and other long-acting stimulants (Adler et al., 2008; Adler, Zimmerman, et al., 2009; Spencer et al., 2007; Weisler et al., 2006), with the most frequently reported TEAEs including insomnia, decreased appetite, dry mouth, decreased weight, and increases in blood pressure. There was a dose–response relationship observed for severe TEAEs and weight decrease−related TEAEs, with higher triple-bead MAS doses being associated with an increased frequency of these TEAEs. Increases in mean pulse were also dose dependent. The safety and tolerability results by randomized treatment group were consistent with those by actual dose, suggesting that the observed dose–response relationship was not an artifact of the titration schedule.
Sleep quality improved based on PSQI score reductions, which is consistent with previous studies of triple-bead MAS (Spencer et al., 2008) and other stimulants (Roth & Zinsenheim, 2009). However, insomnia was the most frequently reported TEAE, the most frequently reported severe TEAE, and the most frequently reported TEAE leading to study discontinuation. There was no apparent dose–response relationship for insomnia-related TEAEs, but the highest frequency of insomnia-related TEAEs was observed with 75 mg triple-bead MAS. In the current study, the frequency of any insomnia-related TEAE (41.7% [including insomnia, initial insomnia, middle insomnia, terminal insomnia, and early morning awakening]) with triple-bead MAS (all doses combined) exceeded the frequency reported for dose-optimized triple-bead MAS (29.2%) (Spencer et al., 2008) and for other long-acting stimulants (osmotic controlled-release oral delivery system methylphenidate [OROS-MPH]: insomnia [9.1%] and initial insomnia [7.3%]; MAS XR: insomnia [21%-30%]; extended-release dexmethylphenidate: insomnia [13%-18.5%]; lisdexamfetamine dimesylate: insomnia [17%-21%])(Adler et al., 2008; Adler, Zimmerman, et al., 2009; Spencer et al., 2007; Weisler et al., 2006). However, it is important to note that at least one of these studies (Adler, Zimmerman, et al., 2009) used a dose-optimization design rather than a forced-dose design, which may have influenced the frequency of reported TEAEs.
The magnitude of the mean blood pressure and pulse changes observed in this study with triple-bead MAS (SBP: −0.3-1.5 mmHg; DBP: −0.3-0.4 mmHg; pulse: 0.9-5.4 bpm) is within the range reported for other long-acting stimulants (OROS-MPH: SBP, −1.2 mmHg; DBP, 1.1 mmHg; pulse, 3.6 bpm; MAS XR: SBP, 0.3-4.3 mmHg; pulse, 4.2-6.2 bpm; extended-release dexmethylphenidate: SBP, −0.5 mmHg; DBP, 1.0 mmHg; pulse, 3.1-6.0 bpm) (Adler, Zimmerman, et al., 2009; Spencer et al., 2007; Weisler et al., 2006). Outlier analyses indicated that the proportion of participants with prespecified changes in SBP, DBP, and pulse at any visit was highest with 75 mg triple-bead MAS.
Some aspects of this study may limit its generalizability. First, the forced-dose titration design does not model flexible-dose treatment strategies used in clinical settings. Second, the study was not powered for assessment of secondary efficacy endpoints; consequently, the nominal p values reported for those endpoints are descriptive. Third, the short treatment duration does not allow for assessment of the long-term safety, tolerability, and effectiveness of triple-bead MAS.
Conclusion
All triple-bead MAS doses assessed (25, 50, and 75 mg) were statistically superior to placebo in reducing ADHD symptoms in adults; however, there was no statistical evidence of a dose–response relationship for efficacy. The short-term safety and tolerability profile of triple-bead MAS was similar to other long-acting stimulants; however, higher rates of insomnia-related TEAEs were observed compared with other long-acting stimulants (Adler et al., 2008; Adler, Zimmerman, et al., 2009; Spencer et al., 2007; Weisler et al., 2006). There was no apparent dose–response relationship for insomnia-related TEAEs, but insomnia was the most frequently reported TEAE, the most frequently reported severe TEAE, and the most frequently reported TEAE leading to study discontinuation.
Footnotes
Acknowledgements
Under the direction of the authors, writing assistance was provided by Stefan Kolata, PhD (a former employee of Complete Healthcare Communications [CHC], Chadds Ford, PA), and Craig Slawecki, PhD (a current employee of CHC). Editorial assistance in the form of proofreading, copyediting, and fact checking was also provided by CHC. Shailesh Desai, PhD, from Shire, reviewed and edited the manuscript for scientific accuracy. The authors acknowledge Colleen Anderson, MEd, for her contributions to the clinical development program for triple-bead MAS and for her insightful comments on this manuscript.
Authors’ Note
Glen Frick is currently employed at Endo Pharmaceuticals, Malvern, PA, USA
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Glen Frick is a former employee of Shire and holds stock and/or stock options in Shire. Brian Yan is an employee of Shire and holds stock and/or stock options in Shire. In the past year, Lenard Adler has received grant/research support from Sunovion Pharmaceuticals, Purdue Pharmaceuticals, Enzymotec, Shire, and Lundbeck; has served as a consultant to Enzymotec, Alcobra Pharmaceuticals, Rhodes Pharmaceuticals, Shire, the National Football League, and Major League Baseball. He has also received royalty payments (as inventor) from New York University for license of adult ADHD scales and training materials since 2004. Lenard Adler has no conflicts in regard to stock ownership or speaker’s bureaus.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This Clinical research was funded by the sponsor, Shire Development LLC (Lexington, MA). Shire provided funding to CHC for support in writing and editing this manuscript.
Author Biographies
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.