
Abstract
Introduction
Psychostimulants are commonly prescribed medications for the treatment of ADHD in adults (Castle, Aubert, Verbrugge, Khalid, & Epstein, 2007). Although long-acting psychostimulant formulations can alleviate symptoms of ADHD for as long as 8 to 14 hr (Adler et al., 2017; Thomas, Rostain, & Prevatt, 2013), some individuals with ADHD may require symptom control beyond 14 hr. With currently available long-acting medications, some adults with ADHD may need to use dose-augmentation strategies (e.g., an extended-release formulation in the morning followed by an immediate-release formulation later in the day) to achieve symptom control into the evening hours (Adler, Reingold, Morrill, & Wilens, 2006).
Triple-bead mixed amphetamine salts (MAS), also known as SHP465, is a once-daily, triple-bead, sustained-release, single-entity mixed amphetamine salt product for oral administration. It is comprised of sulfate salts of d-amphetamine and amphetamine, with d-amphetamine saccharate and amphetamine aspartate monohydrate. The capsule contains three types of drug-releasing beads, which provide for immediate release, pulsatile delayed release, and sustained-release of the MAS. In a Phase 1 study, the pharmacokinetic profile of 37.5-mg triple-bead MAS was similar to that of a morning dose of 25-mg MAS XR supplemented 8 hr later by 12.5-mg MAS IR (Ermer et al., 2007). The short-term efficacy, safety, and tolerability of triple-bead MAS have been demonstrated in two randomized, double-blind studies (Frick, Yan, & Adler, 2016; Spencer, Adler, Weisler, & Youcha, 2008). In a 7-week, Phase 3, dose-optimization study, triple-bead MAS (12.5-75 mg) produced significantly greater reductions in ADHD Rating Scale–IV (ADHD-RS-IV) total score than did placebo and was associated with significantly greater improvement on the Clinical Global Impressions−Improvement (CGI-I) scale than placebo in adults with ADHD (Spencer et al., 2008). In a 6-week, Phase 3, forced-dose titration study, triple-bead MAS (25, 50, and 75 mg) produced significantly greater reductions in ADHD-RS-IV total score than did placebo in adults with ADHD, but a statistically significant dose response for efficacy was not observed across the triple-bead MAS dose range assessed in the study (Frick et al., 2016). Across both short-term Phase 3 studies, the most frequently reported treatment-emergent adverse events (TEAEs) with triple-bead MAS included insomnia, dry mouth, decreased appetite, headache, and decreased weight (Frick et al., 2016; Spencer et al., 2008). Insomnia was the most frequently reported TEAE in both studies (Frick et al., 2016; Spencer et al., 2008), with most insomnia-related TEAEs being mild to moderate in severity and a small percentage leading to study discontinuation (2.2% in the dose-optimized study and 7.7% in the fixed-dose study across all doses; Frick et al., 2016; Spencer et al., 2008). M ± SD vital sign changes with triple-bead MAS from baseline to end-of-study (EOS) in the dose-optimized and fixed-dose (overall across doses) studies were 1.3 ± 9.3 and 0.3 ± 10.48 mmHg, respectively, for systolic blood pressure (SBP); 1.8 ± 6.7 and −0.1 ± 7.43 mmHg for diastolic blood pressure (DBP); and 4.7 ± 11.1 and 3.5 ± 10.33 bpm for pulse (Frick et al., 2016; Spencer et al., 2008).
Long-term outcomes for U.S. Food and Drug Administration (FDA)–approved long-acting psychostimulants in adults with ADHD have been examined in multiple studies (Adler et al., 2011; Adler, Spencer, McGough, Jiang, & Muniz, 2009; Biederman et al., 2005; Weisler et al., 2009). The most frequently reported TEAEs with long-term psychostimulant treatment include insomnia, decreased appetite or anorexia, dry mouth, and headache (Adler et al., 2011; Adler, Spencer, McGough, Jiang, & Muniz, 2009; Biederman et al., 2005; Weisler et al., 2009). Long-term treatment with these agents is also generally associated with increases in blood pressure and pulse (Adler et al., 2011; Adler, Spencer, et al., 2009; Weisler et al., 2009). In terms of ADHD symptom management, open-label extension studies have shown that long-term treatment with FDA-approved psychostimulants maintains the ADHD symptom reductions observed during short-term, placebo-controlled studies (Adler et al., 2011; Adler, Spencer, et al., 2009; Biederman et al., 2005; Weisler et al., 2009).
This report describes the long-term safety and tolerability of triple-bead MAS in participants from the aforementioned Phase 3 studies (Frick et al., 2016; Spencer et al., 2008) who enrolled in a 12-month open-label extension. The primary objective of this study was to evaluate the long-term safety and tolerability of triple-bead MAS in adults with ADHD. The key secondary objective was to assess the long-term clinical outcomes of triple-bead MAS in adults with ADHD based on changes in ADHD-RS-IV total score and improvement on the CGI-I scale.
Method
Study Design and Treatment Regimen
This was a 12-month, Phase 3, multicenter, open-label extension (clinicaltrials.gov registry: NCT00152035) of two Phase 3 studies (clinicaltrials.gov registry: NCT00152022 and NCT00150579). Of the 55 U.S. sites that participated, 54 enrolled participants. The study was conducted from March 10, 2005 to November 7, 2006. Before study initiation, the study protocol was approved by a central institutional review board (IRB) for 48 sites and local IRBs for seven sites. The study was managed by Quintiles, Inc. (San Diego, CA), which was responsible for submitting the protocol and amendments, informed consent documents, relevant supporting information, and all recruitment information to the central IRB for review; the seven sites using local IRBs submitted all study-related documents directly to the IRB. Written informed consent was obtained from all participants after reading written study information and receiving an explanation of the study, including the study objectives, potential benefits and risks, and the participants’ rights and responsibilities. All consent documentation was in accordance with applicable regulations of good clinical practice and guidelines established by the Health Information Portability and Privacy Act of 1996. All investigators received training before study initiation.
The study consisted of 4 weeks of dose optimization, 11 months of dose maintenance, and a 30-day (±5 days) follow-up period. During the study, triple-bead MAS was to be taken in the morning. The original protocol for this study called for an assessment of 12.5-, 25-, 37.5-, 50-, 62.5-, and 75-mg triple-bead MAS. However, results of the antecedent Phase 3 studies demonstrated that optimal triple-bead MAS dosing ranged from 12.5 to 50 mg, with no clear evidence of increased efficacy at doses greater than 50 mg. Furthermore, vital sign data indicated that participants treated with 75-mg triple-bead MAS had the largest mean blood pressure and heart rate increases in the forced-dose study (Frick et al., 2016). Therefore, a protocol amendment modified the maximum allowed dose to be no greater than 50-mg triple-bead MAS. All participants previously optimized to 62.5 or 75 mg had their dose reduced to 50 mg or less. Despite this protocol amendment, this report describes data for all triple-bead MAS doses assessed during the study. Triple-bead MAS capsules were provided in 1-week supplies during dose optimization and in 1-month supplies during dose maintenance.
The last double-blind treatment visit of the antecedent Phase 3 study served as the first visit for this extension study (visit 0; open-label baseline). During dose optimization, all participants began treatment with 12.5- or 25-mg triple-bead MAS, regardless of the maximum triple-bead MAS dosage used during the antecedent study. The triple-bead MAS dosage was increased by 12.5 or 25 mg each week until an optimal dose was reached. Optimal dosing was based on the clinical judgment of the investigator, who took evaluations of TEAEs and vital sign changes into account; titration to lower dosages was permitted in decrements of 12.5 or 25 mg. After reaching an optimized dosage, it was to be maintained until the end of the optimization period, with continued evaluation of safety and efficacy being made at scheduled clinic visits. The assessment window for weekly visits during dose optimization was ±2 days from the expected visit day. During dose maintenance, participants continued treatment at the established optimized dosage. Assessments proceeded for 11 months, with monthly visits (±5 days from the expected visits) scheduled for evaluation and medication disbursement. Increases or decreases in the daily dose (by 12.5 or 25 mg) were allowed during this period based on the investigator’s clinical judgment of safety and efficacy measures. In participants optimized to 62.5- or 75-mg triple-bead MAS before implementation of the protocol amendment, a one-step dose reduction was required to achieve a dosage of ≤50 mg. During the follow-up period, participants were contacted by phone 30 (±5) days after their last study drug dose, and information on new or ongoing adverse events (AEs) that had not resolved was collected. If the principal investigator determined that AEs were not acceptably resolved, follow-up continued until there was no longer a safety concern.
Participants were free to withdraw from the study at any time for any reason, and a participant could be discontinued from the study by the investigator at any time in the interest of a participant’s safety. Reasons for participant withdrawal included the following: withdrawn consent or refusal to continue treatment or follow study procedures; if the participant’s ADHD required alternative treatment or if the participant required concomitant medication disallowed by the study protocol; the occurrence of unmanageable AEs; if the participant became pregnant; or other reasons (e.g., significant protocol violation or noncompliance).
Participants
Eligible adults (18-55 years of age at the time of antecedent study consent) were men or nonpregnant, nonlactating women who met Diagnostic and Statistical Manual of Mental Disorders (4th ed., text rev.; DSM-IV-TR; American Psychiatric Association, 2000) criteria for ADHD and who satisfied all antecedent study entry criteria, including having ADHD-RS-IV total scores of ≥24 (participants from study NCT00150579) or ≥32 (participants from study NCT00152022) at antecedent study baseline. Participants were also required to have had satisfactory medical assessments with no clinically significant abnormalities (as determined by medical history, physical examination, and clinical and laboratory evaluations) and to be able to understand and comply with study procedures and restrictions. Treatment response during the antecedent study was not a prespecified criterion for study inclusion; any participant who completed ≥4 weeks of double-blind treatment in the antecedent studies without experiencing clinically significant AEs that precluded continued treatment was eligible for study inclusion.
Key exclusion criteria included termination from an antecedent study for nonadherence and/or having experienced a serious AE (SAE) or an AE resulting in termination; emergent comorbid psychiatric diagnosis with significant symptoms, such as any severe comorbid Axis I or II disorder (mild to moderate social phobia or dysthymia was not exclusionary), or other symptomatic manifestations that contraindicated triple-bead MAS treatment or could confound study assessments; a lifetime history of psychosis or bipolar disorder; a concurrent chronic or acute illness or unstable medical condition that could confound safety assessments, increase risk, or lead to difficulty complying with the protocol; a history of seizures (other than infantile febrile seizures), any tic disorder, or a current diagnosis and/or family history of Tourette syndrome; having known cardiac structural abnormalities or any other cardiac condition; a clinically significant electrocardiogram (ECG) or laboratory abnormalities at baseline of the current study; use of excluded medications, including antidepressants, selective norepinephrine reuptake inhibitors, antipsychotics, anxiolytics, sedative-hypnotics or sedating antihistamines, psychostimulants or any stimulant- or sympathomimetic-containing preparation, clonidine and guanfacine, monoamine oxidase inhibitors, anticonvulsants, investigational medications, and psychoactive herbal preparations; a documented allergy, intolerance, or history of nonresponse to methylphenidate or amphetamine; and a drug dependence or substance abuse disorder according to DSM-IV-TR criteria (excluding nicotine) or a positive urine drug result at baseline for drugs of abuse.
Endpoints
Safety and tolerability
Safety and tolerability endpoints consisted of AEs, clinical laboratory and vital sign assessments, physical examinations, ECGs, and assessment of sleep with the Pittsburgh Sleep Quality Index (PSQI). AEs were assessed from the time of informed consent until the end of treatment and were categorized by their relatedness to treatment, intensity, and seriousness based on the clinical judgment of the study investigators. Coding of AEs was based on the Medical Dictionary for Regulatory Activities, version 8.1. The final assessment during the antecedent study served as the open-label baseline for vital signs, physical examinations, weight, clinical laboratory assessments, ECG, and the PSQI. Clinical laboratory assessments occurred at the Month 6 and Month 12/EOS visits, where EOS was defined as the last nonmissing visit after open-label baseline. Vital signs were assessed at every study visit starting at the Week 1 dose-optimization visit and at follow-up; a midweek assessment (4 ± 1 days from the prior visit) was required if a participant initiated treatment with 62.5- or 75-mg triple-bead MAS. Twelve-lead ECGs were assessed at Weeks 1 through 5, Months 2 through 6, Month 9, and EOS.
Sleep quality was assessed at every study visit starting at the Week 1 dose-optimization visit using a modified 18-item PSQI. Individual items on the PSQI can be used to generate a global score and seven component scores that assess subjective sleep quality, sleep latency, sleep duration, habitual sleep efficiency, sleep disturbances, use of sleeping medication, and daytime dysfunction (Backhaus, Junghanns, Broocks, Riemann, & Hohagen, 2002; Buysse, Reynolds, Monk, Berman, & Kupfer, 1989). Global scores on the modified PSQI ranged from 0 to 21, with higher scores indicating lower quality of sleep.
Clinical outcomes
The ADHD-RS-IV and CGI-I were used to measure symptom severity and global disease improvement, respectively. The ADHD-RS-IV consists of 18 items designed to assess current ADHD symptomatology; each item is scored on a 4-point scale ranging from 0 (no symptoms) to 3 (severe symptoms); total score ranges from 0 to 54 (DuPaul, Power, Anastopoulos, & Reid, 1998). The ADHD-RS-IV includes two 9-item subscales that assess inattentiveness and hyperactivity/impulsivity, respectively. The ADHD-RS-IV was developed for children; however, with training and the use of prompts, a clinician can use the ADHD-RS-IV to interpret the adult manifestations of ADHD symptoms and fully explore the breadth of those symptoms (Adler & Cohen, 2004; Murphy & Adler, 2004). The use of the ADHD-RS-IV with adult prompts has been shown to be effective in assessing treatment effects in adults with ADHD (Adler et al., 2013; Adler et al., 2008; Spencer et al., 2007; Spencer et al., 2001; Weisler et al., 2009; Wilens et al., 2005). In this study, investigators were provided with and trained on the use of the ADHD-RS-IV with adult prompts using standardized methods (Adler et al., 2005) in a manner consistent with other triple-bead MAS studies (Frick et al., 2016; Spencer et al., 2008). The ADHD-RS-IV was assessed at open-label baseline, at Weeks 1 through 4, and at Months 2 through EOS.
The CGI-I provides a global impression of disease improvement over time (Guy, 1976). Improvement of the participant’s condition was rated on an 8-point scale by the investigator (1 = very much improved, 2 = much improved, 3 = minimally improved, 4 = no change, 5 = minimally worse, 6 = much worse, and 7 = very much worse]); a score of 0 indicates not assessed. Scores on the CGI-I were assessed at every study visit starting at the Week 1 dose-optimization visit; improvement was assessed against Clinical Global Impressions–Severity (CGI-S) scores (range = 1 [normal, not at all ill] to 7 [among the most extremely ill]) at antecedent study baseline.
Data Presentation and Analyses
Safety and tolerability were assessed in the safety population (participants who took ≥1 study drug dose, including those who received medication but were immediately lost to follow-up). Safety and tolerability findings are presented using descriptive statistics. For assessment of PSQI global score, no data imputation was used for missing items.
Clinical outcomes were measured in the intent-to-treat (ITT) population (all participants having ≥1 post-open-label baseline assessment and an antecedent study baseline assessment for ADHD-RS-IV). Changes in ADHD-RS-IV total score and subscale scores from antecedent study baseline and from open-label baseline to EOS were assessed using one-sample t tests, where EOS was defined as the last nonmissing visit after open-label baseline. Hypothesis testing was conducted at the 5% significance level. Scores on the CGI-I were dichotomized as improved (scores of 1 or 2 [very much or much improved]) versus not improved (scores of 3-7 [minimally improved, no change, minimally worse, much worse, and very much worse]) and are presented descriptively.
Results
Disposition and Demographics
All 505 enrolled participants were included in the safety population; 502 participants were included in the ITT population (Figure 1). A total of 239 enrolled participants (47.3%) did not complete the study. Over the course of the study, participants discontinued because of withdrawn consent (n = 73), AEs (n = 66), lost to follow-up (n = 54; after receiving at least two telephone calls and a certified letter), protocol violation (n = 19), noncompliance (n = 13), required alternative therapy (n = 7), pregnancy (n = 3), and other (n = 4).

CONSORT diagram.
The majority of participants in the safety population were White, and more than half were men; most study participants were of the combined ADHD subtype (Table 1). ADHD-RS-IV total and subscale scores at open-label baseline were lower in participants randomized to triple-bead MAS during the antecedent studies than in those randomized to placebo during the antecedent studies (Table 1).
Participant Demographics, Safety Population.
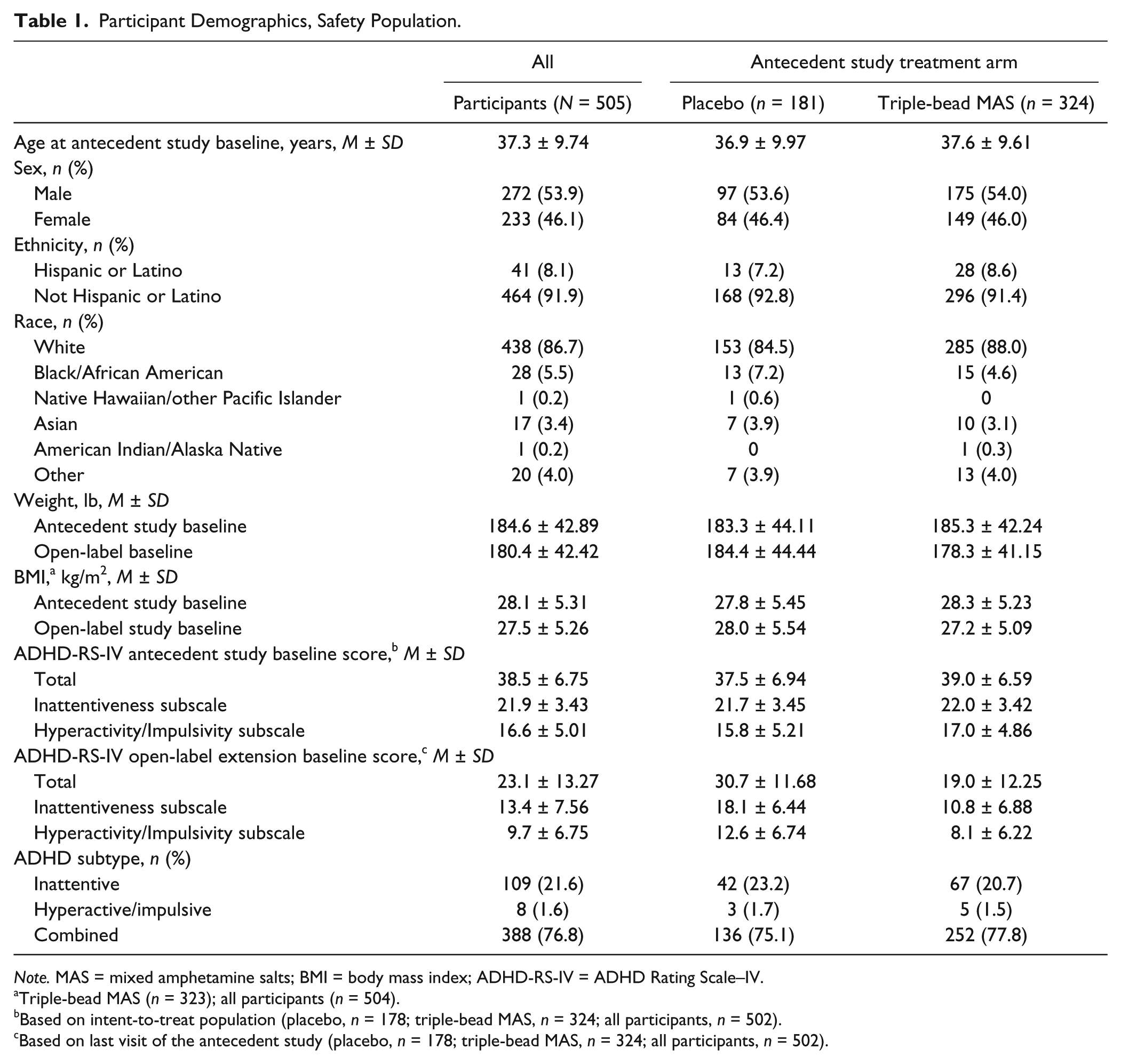
Note. MAS = mixed amphetamine salts; BMI = body mass index; ADHD-RS-IV = ADHD Rating Scale–IV.
Triple-bead MAS (n = 323); all participants (n = 504).
Based on intent-to-treat population (placebo, n = 178; triple-bead MAS, n = 324; all participants, n = 502).
Based on last visit of the antecedent study (placebo, n = 178; triple-bead MAS, n = 324; all participants, n = 502).
Extent of Exposure
During the study, M ± SD exposure to triple-bead MAS was 8.48 ± 4.394 months. A total of 266 (52.7%) participants were exposed to triple-bead MAS for ≥12 months; 362 (71.7%) participants were exposed to triple-bead MAS for ≥6 months. Of the 78 participants exposed to triple-bead MAS for >12 months (12 months was defined as 360 days in the study protocol), most were exposed to triple-bead MAS between 361 and 365 days. Two participants were exposed to triple-bead MAS for 383 and 385 days, respectively. These longer exposure durations were due to difficulties associated with keeping study visits within a 365-day calendar year in the context of real-life clinical practice. At Month 11, the most commonly dispensed dose was 50 mg (48.7%); the M ± SD daily dose during the study was 48.0 ± 15.96 mg (n = 503). The number of participant-years of triple-bead MAS exposure was 350.4 years in the safety analysis set. M ± SD treatment adherence rate, calculated as the number of capsules taken divided by the number of capsules that should have been taken over the treatment period, was 96.4% ± 8.60% for all participants (95.3% ± 11.83% and 97.1% ± 6.03% in participants randomized to placebo or triple-bead MAS, respectively, in the antecedent studies). Mean triple-bead MAS doses and dose changes from the previous visit are reported in Supplemental Table 1. During the dose-maintenance phase, a majority of participants maintained the triple-bead MAS dose established during the dose-optimization phase (range = 76.8% at Month 2 to 92.8% at Month 11).
Safety and Tolerability
AEs
Overall, 89.5% (452/505) of participants experienced a TEAE (Table 2), with the frequency of TEAEs being highest with 75-mg triple-bead MAS (122/158; 77.2%). The frequencies of TEAEs reported by ≥5% of all participants in the safety population are presented in Table 2. The most frequently reported TEAEs (reported by >10% of all participants) were insomnia, headache, dry mouth, upper respiratory tract infection, decreased appetite, weight decreased, and nasopharyngitis. The frequency of TEAEs over the course of the study, which generally decreased over time, is summarized in Supplemental Table 2.
Summary of TEAEs by Triple-Bead MAS Dose at Onset and Overall, Safety Population.
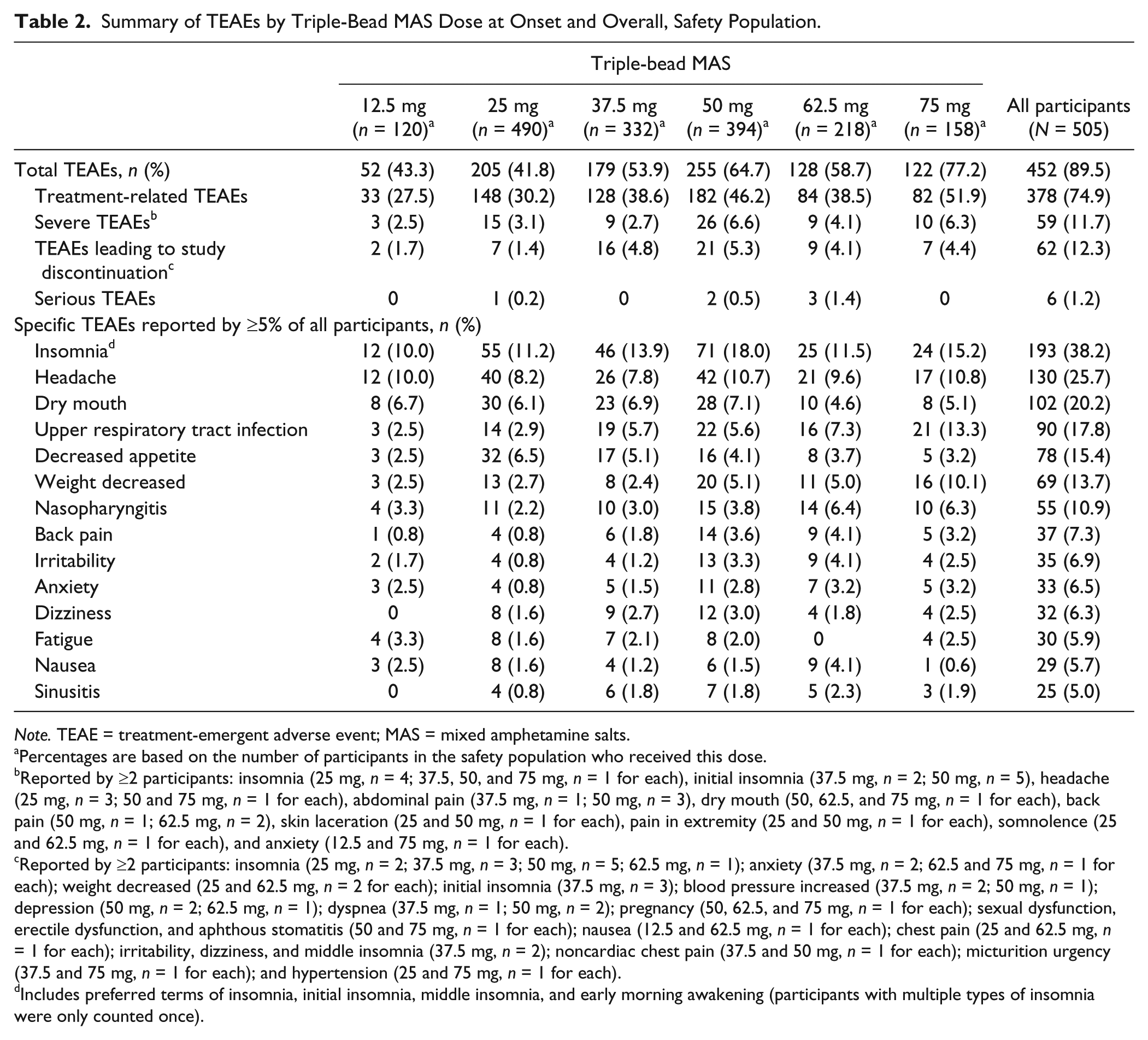
Note. TEAE = treatment-emergent adverse event; MAS = mixed amphetamine salts.
Percentages are based on the number of participants in the safety population who received this dose.
Reported by ≥2 participants: insomnia (25 mg, n = 4; 37.5, 50, and 75 mg, n = 1 for each), initial insomnia (37.5 mg, n = 2; 50 mg, n = 5), headache (25 mg, n = 3; 50 and 75 mg, n = 1 for each), abdominal pain (37.5 mg, n = 1; 50 mg, n = 3), dry mouth (50, 62.5, and 75 mg, n = 1 for each), back pain (50 mg, n = 1; 62.5 mg, n = 2), skin laceration (25 and 50 mg, n = 1 for each), pain in extremity (25 and 50 mg, n = 1 for each), somnolence (25 and 62.5 mg, n = 1 for each), and anxiety (12.5 and 75 mg, n = 1 for each).
Reported by ≥2 participants: insomnia (25 mg, n = 2; 37.5 mg, n = 3; 50 mg, n = 5; 62.5 mg, n = 1); anxiety (37.5 mg, n = 2; 62.5 and 75 mg, n = 1 for each); weight decreased (25 and 62.5 mg, n = 2 for each); initial insomnia (37.5 mg, n = 3); blood pressure increased (37.5 mg, n = 2; 50 mg, n = 1); depression (50 mg, n = 2; 62.5 mg, n = 1); dyspnea (37.5 mg, n = 1; 50 mg, n = 2); pregnancy (50, 62.5, and 75 mg, n = 1 for each); sexual dysfunction, erectile dysfunction, and aphthous stomatitis (50 and 75 mg, n = 1 for each); nausea (12.5 and 62.5 mg, n = 1 for each); chest pain (25 and 62.5 mg, n = 1 for each); irritability, dizziness, and middle insomnia (37.5 mg, n = 2); noncardiac chest pain (37.5 and 50 mg, n = 1 for each); micturition urgency (37.5 and 75 mg, n = 1 for each); and hypertension (25 and 75 mg, n = 1 for each).
Includes preferred terms of insomnia, initial insomnia, middle insomnia, and early morning awakening (participants with multiple types of insomnia were only counted once).
Most TEAEs were mild or moderate in intensity. Severe TEAEs were reported in 59 participants (11.7%), and the most frequently reported severe TEAEs were insomnia-related (Table 2, Footnote b). Serious TEAEs were reported in six participants during the study (25 mg: skin laceration and hand fracture [n = 1]; 50 mg: noncardiac chest pain and dyspnea [n = 1] and testicular torsion [n = 1]; 62.5 mg: third cranial nerve paresis and vision blurred [n = 1], bipolar disorder and suicide attempt [n = 1], and renal cell carcinoma [stage II]/rectal hemorrhage [n = 1]). All serious TEAEs were considered to be unrelated to study medication based on the clinical judgment of the investigators, and all but the case of bipolar disorder resolved. The SAEs of bipolar disorder and suicide attempt occurred in the same participant and resulted in study discontinuation. The participant was diagnosed with bipolar disorder, the symptoms of which stabilized after additional treatment, following hospitalization for the suicide attempt. Sixty-two participants (12.3%) discontinued because of TEAEs (Table 2); the most frequently reported TEAEs leading to discontinuation were related to insomnia (Table 2, Footnote c). The frequency of discontinuation was lower with 12.5- and 25-mg triple-bead MAS than with 37.5-, 50-, 62.5-, and 75-mg triple-bead MAS (Table 2). There were no deaths during the study.
Vital signs (pulse, SBP, and DBP)
Pulse, SBP, and DBP analyses are summarized in Table 3. Mean pulse increased from antecedent study baseline to EOS among all participants and for each triple-bead MAS dose. Mean SBP and DBP increased from antecedent study baseline to EOS among all participants and for all triple-bead MAS doses, except for 62.5-mg triple-bead MAS for SBP and DBP and 25-mg triple-bead MAS for DBP, for which decreases were observed. For mean changes in pulse and blood pressure, there were no apparent relationships or differences based on antecedent study treatment.
EOS and Changes at EOS (M ± SD) in Vital Signs, ECG Parameters, Weight, and BMI by Triple-bead MAS Dose.
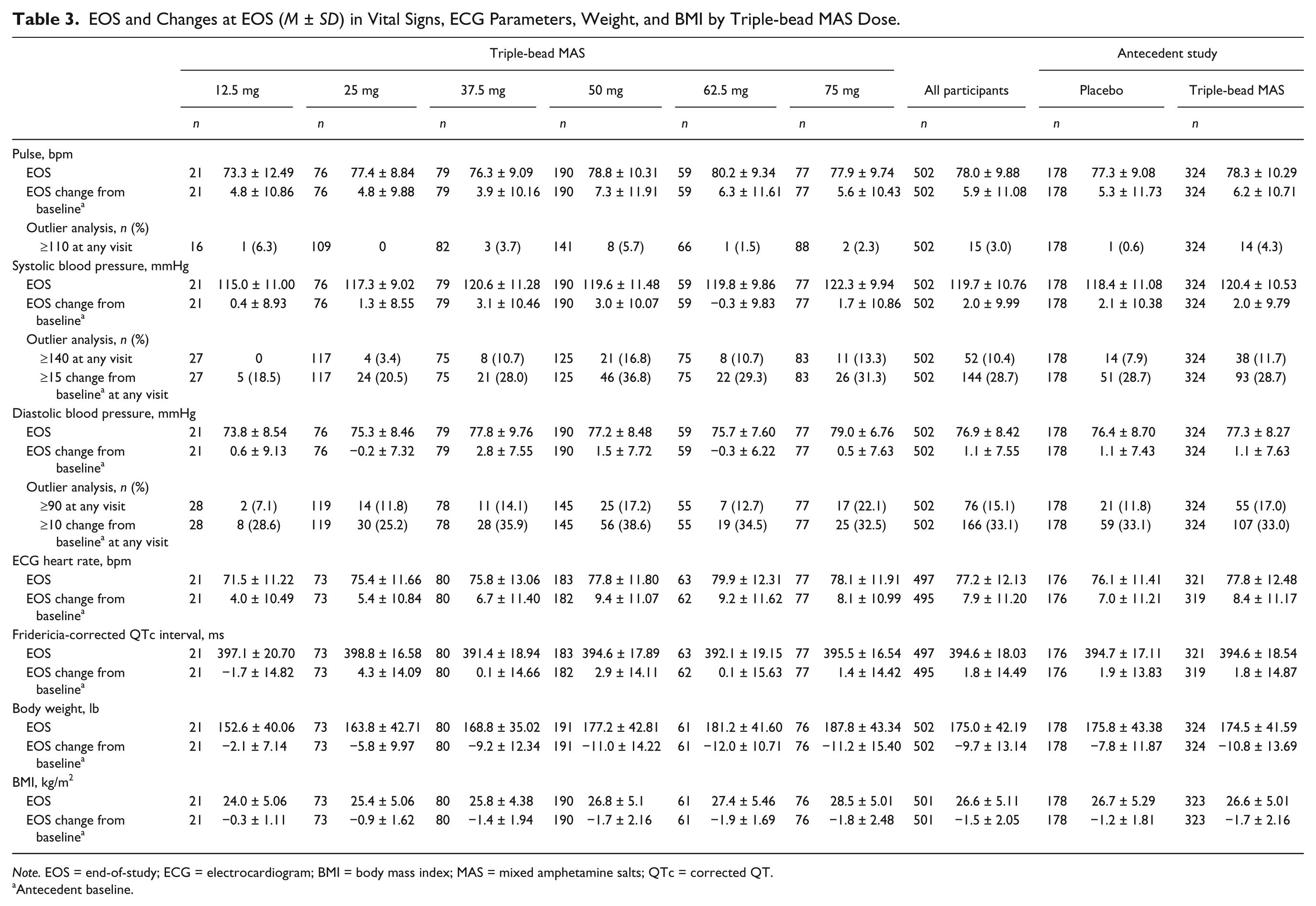
Note. EOS = end-of-study; ECG = electrocardiogram; BMI = body mass index; MAS = mixed amphetamine salts; QTc = corrected QT.
Antecedent baseline.
Outlier blood pressure analyses indicated that the proportion of participants with prespecified changes in SBP (≥140 mmHg or increase ≥15 mmHg from baseline) and DBP (≥90 mmHg or increase ≥10 mmHg from baseline) at any visit was highest at doses ≥37.5-mg triple-bead MAS; however, there was no clear dose–response relationship. A total of 26 participants had SBP ≥140 mmHg and/or DBP ≥90 mmHg and an increase ≥5 mmHg from baseline at two or more consecutive visits. Of these participants, blood pressure returned to normal levels at EOS in 13 cases, remained elevated at EOS in 10 cases (25 mg, n = 2; 37.5 mg, n = 2; 50 mg, n = 5; 62.5 mg, n = 1), and resulted in study discontinuation in three cases (Table 2, Footnote c). Outlier pulse rate analyses indicated that the proportion of participants with prespecified pulse changes (≥110 bpm at any visit) was highest for 50-mg triple-bead MAS (Table 3), with no clear dose–response relationship observed. Of the 15 instances of pulse ≥110 bpm during the study, most were isolated and returned to normal while on treatment.
ECGs, physical examination findings, and clinical laboratory tests
Heart rate based on the ECG increased with triple-bead MAS at EOS compared with antecedent study baseline (Table 3), with larger magnitude changes observed with higher triple-bead MAS dosages (50, 62.5, and 75 mg) than with lower dosages (12.5, 25, and 37.5 mg). Changes in Fridericia-corrected QTc interval (QTcF) were not deemed clinically relevant; there were no participants with QTcF ≥500 ms at EOS or at any point during the study.
During the course of the study, participants’ M ± SD body weight and body mass index decreased (Table 3), with larger magnitude changes observed with higher triple-bead MAS doses (50, 62.5, and 75 mg) than with lower doses (12.5, 25, and 37.5 mg). Laboratory test parameters were minimally changed during treatment with triple-bead MAS, with small mean changes, most of which were not considered to be clinically significant.
PSQI total scores
Mean PSQI total score changes indicated that overall sleep quality improved during the study. At the antecedent and open-label baselines, M ± SD baseline scores were 6.9 ± 3.06 (n = 505) and 5.3 ± 3.21 (n = 504), respectively, in all participants; 7.1 ± 2.98 (n = 181) and 5.5 ± 3.35 (n = 180) in those randomized to placebo during the antecedent studies; and 6.9 ± 3.11 (n = 324) and 5.1 ± 3.12 (n = 324) in those randomized to triple-bead MAS during the antecedent studies. M ± SD changes in PSQI total score from antecedent study baseline at Month 12 were −2.3 ± 3.48 (n = 266) in all participants (−1.9 ± 3.41 and −2.4 ± 3.52 in those randomized to placebo [n = 85] and triple-bead MAS [n = 181], respectively, during the antecedent studies).
Clinical Outcomes
ADHD-RS-IV
Figure 2 depicts mean ADHD-RS-IV total scores from baseline of the antecedent study through EOS in all participants and by antecedent study treatment assignment; baseline values are summarized in Table 1. At open-label baseline (which is the same as the last visit of the antecedent study), mean ADHD-RS-IV total scores were higher in participants randomized to placebo than to triple-bead MAS during the antecedent studies (Table 1). From treatment Week 4 (after the end of dose optimization) through EOS, ADHD-RS-IV total scores were roughly comparable between participants randomized to placebo or triple-bead MAS during the antecedent studies. At EOS, M ± SD reductions from antecedent study baseline in ADHD-RS-IV total scores were observed in all participants in the ITT population, in participants randomized to placebo during the antecedent studies, and in participants randomized to triple-bead MAS during the antecedent studies (all nominal p < .0001 vs. antecedent and open-label baselines; Figure 2 and Table 4). Changes in ADHD-RS-IV total score from antecedent study baseline at EOS between men and women or by ADHD subtype were similar. Similarly, score reductions on the ADHD-RS-IV Inattentiveness and Hyperactivity/Impulsivity subscales from antecedent study and open-label baselines at EOS were nominally significant (all nominal p < .0001; Table 4).
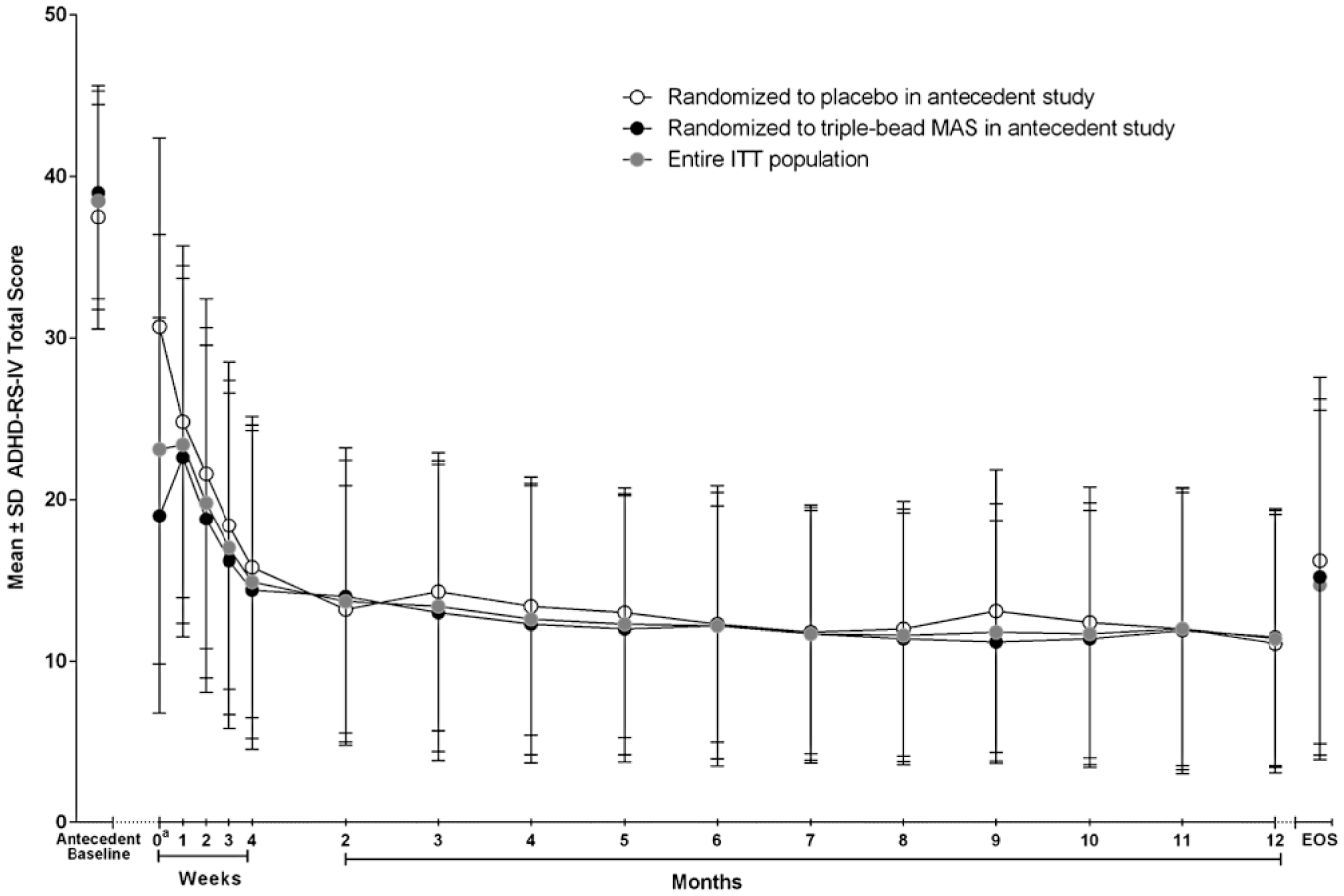
M ± SD ADHD-RS-IV total scores from baseline of the antecedent studies at each assessment, ITT population.
M ± SD ADHD-RS-IV Total and ADHD Subscale Score Changes in the ITT Population.
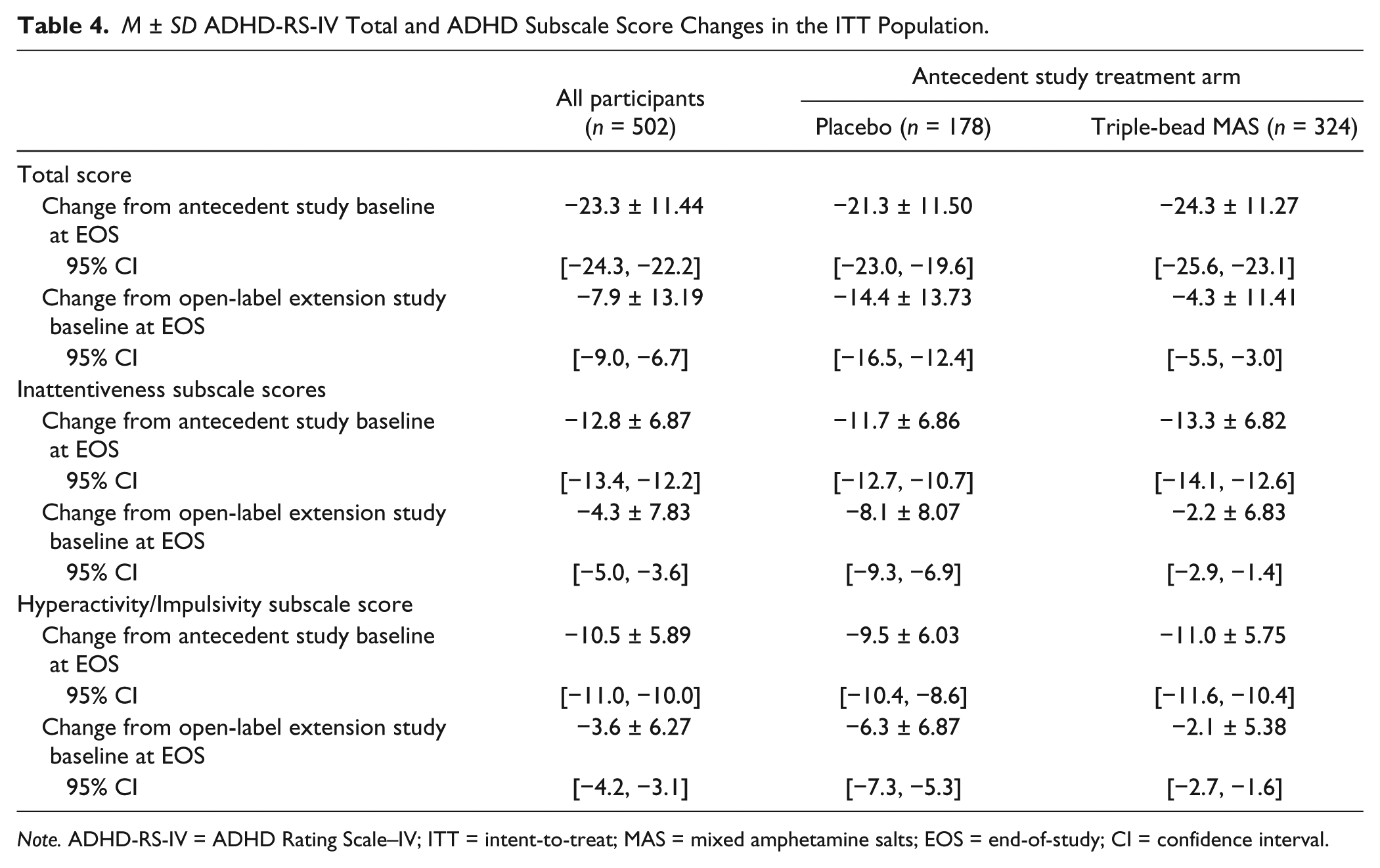
Note. ADHD-RS-IV = ADHD Rating Scale–IV; ITT = intent-to-treat; MAS = mixed amphetamine salts; EOS = end-of-study; CI = confidence interval.
Dichotomized CGI-I
At open-label extension baseline, 52.0% (261/502) of all participants in the ITT population, 21.3% (38/178) of those randomized to placebo in the antecedent studies, and 68.8% (223/324) of those randomized to triple-bead MAS in the antecedent studies were categorized as improved on the CGI-I relative to the antecedent study baseline. At EOS, 79.3% (398/502) of all participants in the ITT population (73.0% [130/178] of those randomized to placebo in the antecedent studies and 82.7% [268/324] of those randomized to triple-bead MAS in the antecedent studies) were categorized as improved on the CGI-I (reported scores of 1 [very much improved] or 2 [much improved]) relative to antecedent study baseline.
Discussion
The long-term safety and tolerability profile of triple-bead MAS in this study was generally comparable with the profiles observed for FDA-approved long-acting psychostimulants in terms of TEAEs, cardiovascular effects, and weight (Adler et al., 2011; Adler, Spencer, et al., 2009; Biederman et al., 2005; Weisler et al., 2009). The most frequently reported TEAEs (>10% of all participants) were insomnia, headache, dry mouth, upper respiratory tract infection, decreased appetite, weight decreased, and nasopharyngitis; most TEAEs were mild to moderate in severity. The most frequently reported severe TEAEs and the most frequently reported TEAEs that resulted in study discontinuation were related to insomnia. Although occurrence of bipolar disorder as a serious TEAE was considered to be unrelated to treatment by the study investigator, it cannot be stated with certainty that it was not related to treatment because stimulants can be associated with the induction of mania (Goldsmith, Singh, & Chang, 2011).
Triple-bead MAS was also associated with increases in blood pressure and pulse. Three participants discontinued because of increased blood pressure; all instances resolved when treatment was terminated. Although there was no clear dose–response relationship observed for safety and tolerability assessments, higher frequencies of TEAEs and higher magnitude changes in vital signs and body weight were generally observed with higher triple-bead MAS doses. Outlier analyses also indicated that the proportion of participants with prespecified changes in SBP and DBP at any visit was highest for triple-bead MAS doses ≥37.5 mg. As is recommended with all psychostimulants (Adler, Weisler, Goodman, Hamdani, & Niebler, 2009), there is a need to monitor blood pressure during treatment with triple-bead MAS.
The long-term safety and tolerability findings described in this report are generally consistent with short-term effects of triple-bead MAS observed in the antecedent studies (Frick et al., 2016; Spencer et al., 2008). In addition, the magnitude of the mean blood pressure and pulse changes observed with triple-bead MAS in the current study (SBP, −0.3 to 3.1 mmHg; DBP, −0.3 to 2.8 mmHg; pulse, 3.9-7.3 bpm) was generally within the ranges reported for FDA-approved long-acting stimulants (osmotic controlled-release oral delivery system [OROS]- methylphenidate: SBP, 2.6 mmHg; DBP, 1.9 mmHg; pulse, 4.1 bpm; extended-release dexmethylphenidate: SBP, 2.3 mmHg; DBP, 1.6 mmHg; pulse, 3.7 bpm; lisdexamfetamine dimesylate [LDX]: SBP, 3.1 mmHg; DBP, 1.3 mmHg; pulse, 3.2 bpm) in long-term safety studies (Adler et al., 2011; Adler, Spencer, et al., 2009; Weisler et al., 2009). The number of participants with SBP ≥140 mmHg (10.4%) or DBP ≥90 mmHg (15.1%) at any point during the study was slightly higher than similar values reported in long-term safety studies for LDX (SBP or DBP, 9.2%) and OROS-methylphenidate (SBP, 9.6%; DBP, 12.0%; Adler et al., 2011; Weisler et al., 2009).
Overall sleep quality tended to improve based on the numerical reductions in PSQI total score at Month 12 with triple-bead MAS, an effect also seen in long-term safety studies of LDX in adults with ADHD (Weisler et al., 2009) and in the antecedent studies for this long-term safety study (Frick et al., 2016; Spencer et al., 2008). However, insomnia was the most frequently reported TEAE, the most frequently reported severe TEAE, and the most frequently reported TEAE leading to study discontinuation. Furthermore, the frequency of any type of insomnia-related TEAEs with triple-bead MAS in the current study (38.2% [includes preferred terms of insomnia, initial insomnia, middle insomnia, and early morning awakening]) exceeded the frequencies reported in long-term safety studies of FDA-approved long-acting stimulants (MAS XR, 18.3%; OROS-methylphenidate, 20.7%; LDX, 19.5%; extended-release dexmethylphenidate, 20.0%; Adler et al., 2011; Adler, Spencer, et al., 2009; Biederman et al., 2005; Weisler et al., 2009). Although the improvement in sleep based on PSQI total score reductions at Month 12 appears to conflict with the overall frequency of insomnia-related TEAEs reported during the study, the low frequency of insomnia-related TEAEs at Month 12 (4 [1.4%]; see Supplemental Table 2) is more consistent with an overall lack of sleep impairment at EOS.
Triple-bead MAS demonstrated improved long-term clinical outcomes for ADHD symptoms for up to 12 months, as measured by reductions in ADHD-RS-IV total score. Participants randomized to triple-bead MAS during the antecedent studies maintained symptom reductions observed during short-term treatment, and participants randomized to placebo during the antecedent studies exhibited ADHD symptom reductions compared with antecedent and open-label study baseline. Reductions in ADHD-RS-IV total score were observed in men and women and across all ADHD subtypes, with no notable differences in the magnitude of score reductions. Consistent with these findings, score reductions on the ADHD-RS-IV Inattentiveness and Hyperactivity/Impulsivity subscales and improvement on the CGI-I are also supportive of the view that long-term triple-bead MAS treatment is associated with improved clinical outcome in adults with ADHD. These overall findings are consistent with the reported effects of FDA-approved long-acting psychostimulants (Adler et al., 2011; Adler, Spencer, et al., 2009; Biederman et al., 2005; Weisler et al., 2009).
The findings of this open-label study should be considered in light of potential limitations. First, the study was not designed to assess dose–response relationships of triple-bead MAS in regard to safety and tolerability endpoints, so drawing inferences from these data should be done with caution. Second, this was an open-label study that was not powered for statistical assessment of the clinical outcome endpoints, and there was no placebo control group. Therefore, interpretation of the reported nominal p values should be done with caution. The potential influence of selection bias should also be considered because treatment response during the antecedent double-blind studies was not a criterion for participation in this extension study, which would limit the applicability of these findings in individuals who start treatment with triple-bead MAS.
Conclusion
In this 12-month open-label extension, triple-bead MAS exhibited a long-term safety and tolerability profile that was comparable with the profiles observed for other long-acting psychostimulants (Adler et al., 2011; Adler, Spencer, et al., 2009; Biederman et al., 2005; Weisler et al., 2009), with the most frequently reported TEAEs (>10% of all participants) being insomnia, headache, dry mouth, upper respiratory tract infection, decreased appetite, weight decreased, and nasopharyngitis. Triple-bead MAS was also associated with increases in blood pressure and pulse that were within ranges reported for other long-acting stimulants. In addition, triple-bead MAS demonstrated long-term effectiveness in the treatment of ADHD symptoms for up to 12 months, as evidenced by continued reductions in ADHD-RS-IV total score relative to antecedent-study and open-label baselines and by the percentage of participants categorized as improved on the CGI-I at EOS.
Footnotes
Acknowledgements
Shire Development LLC provided funding to Complete Healthcare Communications, Inc. (CHC; Chadds Ford, PA) for support in writing and editing this manuscript. Under the direction of the authors, writing assistance was provided by Stefan Kolata, PhD (a former employee of CHC), and Craig Slawecki, PhD (a current employee of CHC). Editorial assistance in the form of proofreading, copyediting, and fact checking was also provided by CHC. Shailesh Desai, PhD, from Shire, reviewed and edited the manuscript for scientific accuracy. The authors would like to acknowledge Colleen Anderson, MEd, for her contributions to the clinical development program for triple-bead MAS and for her insightful comments on this manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: In the past year, Lenard A. Adler has received grant/research support from Sunovion Pharmaceuticals, Purdue Pharmaceuticals, Enzymotec, Shire Pharmaceuticals, and Lundbeck, and has served as a consultant to Enzymotec, Alcobra Pharmaceuticals, Rhodes Pharmaceuticals, Shire Pharmaceuticals, the National Football League, and Major League Baseball. He has also received royalty payments (as inventor) from NYU for license of adult ADHD scales and training materials since 2004. Lenard A. Adler has no conflicts in regard to stock ownership or speakers bureaus. Glen Frick is a former employee of Shire and holds stock and/or stock options in Shire. Brian Yan is an employee of Shire and holds stock and/or stock options in Shire.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This clinical research was funded by the sponsor, Shire Development LLC (Lexington, MA).
Author Biographies
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.