
Abstract
Background
With the recent Food & Drug Administration (FDA) approval of cellular therapy that requires product manipulation prior to administration in combination with a short stability window, the need was identified for local dose preparation within the pharmacy rather than the off-site stem cell processing laboratory. This approval gave rise to assessment of regulatory standards surrounding cellular therapy, evaluation and revision of current standard operating procedures and policies with formal process validation, assessment of occupational exposure mitigation and safety considerations, and development of staff training and education.
Objective
To describe and provide insight into the stepwise process of FACT validation and onboarding of commercially available cellular therapy products that require sterile compounding manipulation within a pharmacy prior to administration.
Discussion
A multidisciplinary effort is required to attain FACT certification and implement pharmacist compounding of cellular therapy products. 1 Local preparation within a pharmacy facilitates a sound operational workflow and provides a pathway to perform aseptic manipulations of cellular therapy products safely and efficiently.
Conclusion
Safe and successful administration of cellular therapies handled and compounded by pharmacy department staff along with program validation requires a preemptive review utilizing a multidisciplinary approach for process development. This manuscript will provide a foundation based on consistency and transparency in effective cellular therapy sterile compounding and aseptic manipulation, proper handling and disposal procedures, increased communication through creation and optimization of treatment plans and order-sets, standardized medical center staff education, and development of policies and standard operating procedures for the entire health care team.
Keywords
The University of California, San Diego Health System (UCSDH), is comprised of three campuses with a combined capacity of 1101 beds, which includes a blood and marrow transplant unit and medical oncology floor. UCSDH Moores Cancer Center is a National Comprehensive Cancer Network (NCCN) and National Cancer Institute (NCI) comprehensive cancer care center, at which more than 5200 new patients enter into care each year. We anticipated that as a regional specialty center for many rare diseases, there would be an influx of patients in need of cellular therapy products once FDA approved and the simultaneous need for healthcare staff to be trained accordingly. The first commercial autologous cellular therapy to be handled by our Department of Pharmacy was lisocabtagene maraleucel. This drug and others in the pipeline initiated the need for standardized process development.
Assessment of regulatory standards surrounding cellular therapy
Given the lack of regulatory standards and translational application of those standards currently available, there is a need for standardized guidance regarding the safe preparation and handling of cellular therapy for all healthcare personnel who are involved in the care of patients receiving these treatment modalities. This concept applies to both investigational products and FDA approved products. Additionally, the processes and procedures associated with cellular therapy handling, preparation, administration, and patient/staff education must include pharmacy department support and involvement. To ensure compliance and preparedness from a regulatory and safety perspective, standard operating procedures (SOPs), and best practices are needed. 2 Nationally accepted guidance standards are instrumental in providing the foundation for local procedure and policy development.
United States Pharmacopoeia (USP) General Chapter <800> - Hazardous Drugs – Handling in Healthcare Settings is a federally enforceable standard that became compendial applicable on November 1, 2023. 3 The purpose of USP <800 > is to ensure safe handling of hazardous medications to minimize the risk of exposure to healthcare personnel, patients, and the environment. This enforceable standard applies to all healthcare personnel who handle hazardous medication preparations and all entities that receive, store, transport, dispense, administer, or dispose of hazardous medications. Novel therapies including cellular therapy products are not included in USP <800 > and a need exists to develop safe practices for healthcare professionals.
The National Institute for Occupational Safety and Health (NIOSH) develops a list of hazardous drugs in healthcare settings for institutions to use as guidance to prevent occupational exposure. 4 The current and draft version of the list do not include cellular therapy medications, nor an explanation of what standard precautions should be used for handling and administering these preparations. The 6th edition of Biosafety in Microbiological and Biomedical Laboratories (BMBL) from the Centers for Disease Control and Prevention states that genetically altered products pose an occupational risk and a potential health risk to healthcare professionals. 5 These standards and references demonstrate that a regulatory gap exists between cellular therapies in the pipeline of the drug development process and what to anticipate when a drug comes to market with this modality.
The pharmacist's role
As with non-cellular therapies, it is imperative to have pharmacy department support and involvement in the planning, process development and implementation of the medication use process for new medications. Regardless of institution size, through a multi-disciplinary team approach, pharmacists add to the continuum of care through the provision of drug monograph creation, risk assessment evaluation, treatment plan development, policy and procedure creation, and guidance regarding supportive care and adverse event management. Additionally, pharmacists and pharmacy technicians perform the sterile compounding of cellular products that require manipulation to achieve the final dose.
Drawing from previous experience
Evaluation of previous experiences handling the same or similar products, prior to FDA approval, through an investigational pharmacy service may prove useful when developing the process pathway for handling, preparing, and dispensing newly approved cellular therapy products. While investigational therapy protocols are provided on a much smaller scale, these foundational aspects may be identified, reviewed, and adapted to standard non-investigational pharmacy practices. When preparing for use of FDA approved products, factors to consider include transition from investigational to commercial product use to avoid treatment gaps, vetting and approval of formulary addition through the institutional Pharmacy and Therapeutics (P&T) committee, dispensing requirements (such as a risk evaluation and mitigation strategy (REMS) program), product acquisition and mechanisms for prescribing and documentation of dose administration (EHR orders and treatment plans). Additionally, specific techniques for dose handling, preparation and administration may be extrapolated from clinical trial procedures and adapted for use in the similar but transformed non-investigational environment.
Occupational exposure mitigation and safety considerations
The first cellular therapy product to go through the validation process was lisocabtagene maraleucel. An institutional assessment of risk was performed to determine the hazardous category designation. Since this medication is not included on the NIOSH list, the risk assessment was used to determine containment strategies and work practices.3,4 Lisocabtagene maraleucel was evaluated based on the route, formulation, packaging, mechanism of action, possible adverse effects, the level of risk of exposure throughout the medication use process and required manipulation of the product. As required by USP and the California State Board of Pharmacy, our institution maintains a master formulation record of all compounded sterile products, a detailed entry for lisocabtagene was developed that included all the steps of preparation that occur in the negative pressure room.6,7
The AHFS pharmacologic therapeutic class of lisocabtagene maraleucel is 26:12 Gene Therapy, 10:00 Antineoplastic Agents and its mechanism of action has a downstream impact on cells including cell expansion, differentiation, and the lysis of CD-19-positive cells.8,9 The material safety data sheet (MSDS) states that the product should be handled in a Biosafety Level 2 (BSL-2) area 10 and cross references the BMBL for standard microbiological practices involving the containment of BSL-2 related modalities. The MSDS also states that all procedures involving the manipulation of infectious materials that may generate an aerosol should be conducted within a Biological Safety Cabinet (BSC) or other certified physical containment devices. The product must be manipulated using aseptic technique and involves extracting the cells out of a vial where there is a possibility of aerosol generation. Additional personal protective equipment includes the use of eye shields while handling the preparation.
NIOSH has developed a hierarchy of controls, a key method of protecting staff in controlling exposures to potential occupational hazards. 11 There are five controls in the hierarchy which include elimination, substitution, engineering controls, administrative controls, and personal protective equipment (PPE). Elimination and substitution principles involve physically removing the hazard and replacing the hazard, respectively. Engineering controls involve isolating staff from the hazard, which include primary and secondary engineering controls that pharmacy staff can access while performing sterile compounding activities. As an example, BSCs are utilized in the negative pressure rooms of a cleanroom suite to help with containment and hazardous drug exposure mitigation. Administrative controls involve policies and procedures, and personal protective equipment protects staff with appropriate clothing while handling medications. While nurses and physicians have access to administrative controls and personal protective equipment, they traditionally do not have access to primary engineering controls that are externally vented and exhausted to outside air per USP < 800 > standards. Because engineering controls are standardly part of an inpatient pharmacy, there is a notable difference in terms of what colleagues can utilize when considering the aseptic manipulation of lisocabtagene maraleucel and hazardous drug mitigation strategies.
According to NIOSH's safe handling recommendations while working with hazardous drugs, a containment primary engineering control (C-PEC) should be used for withdrawing an intravenous solution from a vial. 4 In addition to possible aerosol generation, other potential means of exposure to lisocabtagene maraleucel include potential residue on outer container(s), withdrawing injectables from parenteral containers, moving the product throughout the medication use process, handling of patient body fluids, and spills. It is the combination of these aforementioned factors involving the properties of the medication itself, containment verbiage as recommended by the MSDS, and NIOSH's recommendations for appropriate aseptic manipulations where lisocabtagene maraleucel was classified by our institution as an anti-neoplastic hazardous drug.
Another consideration involving lisocabtagene maraleucel is the container used to transport and temporarily store cellular products. This special container, referred to as a Dewar, stores cryogens and contains vapor phase liquid nitrogen. Handling products that are encased in vapor phase liquid nitrogen also requires a set of standard precautions that need to be followed to ensure staff safety as it can reach temperatures as low as minus 196 degrees Celsius. As described by Geraghty, R et al., 12 liquid nitrogen can cause frostbite, cold burns, and even death by asphyxiation; precautions and personal protective equipment are essential. Utilizing a Dewar requires the use of cryogenic gloves, which are designed for working in temperatures below minus 80 degrees Celsius. 13 As per the MSDS for lisocabtagene maraleucel, protective eye shields must also be worn while handling this product. Throughout the process of both transferring the product into the cleanroom suite and subsequent aseptic manipulations, pharmacists are expected to ensure all involved staff members adhere to the proper precautions and don the appropriate personal protective equipment. This process for pharmacy teams can be novel and unfamiliar as practice sites typically do not need to utilize Dewars or work with liquid nitrogen. Hence, the development of standardized operating procedures and staff education regarding these precautions is imperative.
In terms of working in a negative pressure room, multiple quality measures are in place to prepare the cleanroom before preparing every cellular therapy product. Before the cells are retrieved from the blood bank using a two-step dual verification process, the temperature and humidity of the negative pressure room and cleaning of the biological safety cabinet are confirmed. The BSCs used for product preparation are deactivated and decontaminated both before the cells enter the negative pressure room and deactivated and decontaminated again after cell preparation to ensure that any subsequent compounded sterile preparation made afterwards has an appropriately cleaned workspace. Regarding spill mitigation and preparedness, spill kits are located in both the receiving area of the inpatient pharmacy and inside the negative pressure room where the aseptic manipulations of lisocabtagene maraleucel take place as per USP <800 > . If a vial or syringe is dropped while performing an aseptic manipulation, there is less of an exposure risk if this scenario were to happen inside a of BSC or C-PEC inside a negative pressure room versus at bedside without any engineering controls.
Evaluation and development of standard operating procedures
To prepare and administer cellular therapy products to patients, institutions must achieve validation of the process via the Foundation for the Accreditation of Cellular Therapy (FACT) certification requirements. 1 The initial process involved gathering key collaborators from all disciplines including pharmacists, nurses, physicians, stem cell processing laboratory personnel and quality assurance experts. Additional committees enlisted for consultation from the pharmacy department included the compounding compliance committee and the hazardous drug committee to assist with cellular therapy product classification, assessment of risk, aseptic manipulation considerations, approach to primary engineering control cleaning, and spill management considerations. The validation elements of the implementation are described in Table 1, with various components of workflow, standard operating procedures, staff qualifications, facility qualification, and equipment and process validation.
Cellular therapy validation process via FACT certification requirements.

An essential part of both FACT accreditation and site certification by the manufacturer is the creation of a standard operating procedure (SOP) that covers the drug product in all aspects of use. 1 While the health system and blood and marrow transplant program had long-standing policies and procedures related to stem cell transplant and cellular therapy, the Department of Pharmacy was required to have a departmental policy as well. To create an SOP focused on cellular therapy products, the pharmacist team gathered current policies that were applicable or adjacent to the cellular therapy process. This included policies regarding sterile compounding, hazardous drugs, occupational exposure, infection control, spill management, personnel training, and environmental health and services. The existing policies were used as references and were linked to the new cellular therapy policy. The SOP also incorporated many facets of USP <800 > to capture how these products would move through the health system. This includes the product receipt in the blood bank, subsequent transport to the inpatient pharmacy cleanroom suite, product preparation, waste and disposal in the cleanroom, transport to the patient care unit, administration, and waste disposal on the patient care unit. The final version of the SOP was reviewed and approved by the Department of Pharmacy leadership, the FACT quality coordinator and Blood and Marrow Transplant (BMT) division chairs, and the manufacturer of lisocabtagene.
Process validation
The Blood and Marrow Transplant Program is accredited by FACT guided by cell therapy standards. The standards provide guidelines that span the entire spectrum of cellular therapy, from donor selection to collection in an apheresis center or operating room, followed by simple or complex processing procedures, and then storage, shipping and labeling of the product, to finally administration and subsequent therapy-related care of the patient, including management and monitoring of toxicities and long-term outcomes. 1 FACT requires that critical processes be validated; this mirrors a Failure Mode and Effects Analysis (FMEA) process as the FACT validation included a team facilitator (the FACT quality coordinator) and specialized team members, a description of the process including potential errors or gaps, an assessment of potential errors and solution creation, and finally a measure of the success of the new program by enacting several dry-runs with a debrief after each. 14 Additionally, facilities providing critical services, including preparation of products for administration must be qualified. 1 Cellular therapy products prepared for administration by the Department of Pharmacy using a thaw and dose method were validated by a multidisciplinary team of experts. This hybrid validation of the process and qualification of facility (inpatient and IDS Pharmacy) was performed collaboratively in a 2-phased approach; prospective as a dry-run using empty product vials supplied by the manufacturer and concurrent using a commercial product for infusion. The IDS pharmacist specialist was included in both the facility qualification and process validation as some cellular therapy products do not meet manufacturer quality assurance release specifications and so are prepared for administration by the IDS pharmacist. The process, however, is mirrored using chain of custody, chain of identity, thaw, dose and distribution documents and procedures.
The team collaborated to develop validation and qualification plans that were approved prior to official testing. A facility qualification checklist was utilized to ensure compliance with FACT facility standards. A process test-subject validation checklist was used to ensure the team followed all critical steps in the process to meet pre-determined acceptance criteria. A new component of the process validation included the use of the transport containers from the negative pressure room to the BMT unit. Because lisocabtagene must be kept at room temperature (20°−25°C) after preparation, the stem cell processing laboratory had to validate a room temperature cooler for transport.
Facility qualification included verification of the following elements: location size, space design, controlled storage areas to prevent mix-ups or contamination, lighting, ventilation, access to sinks, temperature and humidity monitoring, biosafety cabinet cleaning, equipment preventative maintenance, use of appropriate personal protective equipment, trained staff, and SOPs.
Process validation included verification of the following parameters: product transportation, temperature monitoring, chain of custody, chain of identity, accompanying infusion related documentation, availability and use of appropriate personal protective equipment, medication preparation room set-up, two-person label verification, product inspection (integrity prior to thaw), dosing of product per manufacturer request for infusion, final product container label verification, infusion documentation, pre-infusion patient and product verification against orders and accompanying documentation, infusion equipment, availability of provider prior to infusion, verification of availability of 2 doses of tocilizumab, product inspection (integrity), infusion record, patient monitoring, adverse event reporting and disposal of packaging/product containers.
Upon quality review of checklists, test subjects and accompanying documentation, it was noted that no significant problems were encountered, however meticulous notes taken during testing helped with SOP refinement and improvement.
Pharmacy personnel training
While the BMBL and FDA documents provide some guidance, pharmacy department staff must be trained regarding cellular therapy products and the nuances of handling, thawing, and preparing these medications. Before the implementation of lisocabtagene maraleucel, our inpatient pharmacy team had extensive experience with hazardous drug compounding and aseptic manipulation. Additionally, as cellular therapies are considered medications by the FDA, at our institution all CART products require that a pharmacist label the drug prior to infusion with an EHR label. The manipulation of cells, however, was an area of practice that only our investigational drug service pharmacy team had familiarity with. The incorporation of prior experience from the investigational drug service pharmacists added an additional layer of training that was utilized to cover practical aspects of cell manipulation. A small group of pharmacists were selected for participation in the pharmacy department training and validation program. The group consisted of a sterile compounding specialist, blood and marrow transplant/immune effector cell specialist, and an investigational drug service specialist; all were board certified in their respective areas. An additional oncology pharmacist with sterile compounding experience was identified to serve as back-up personnel if a primary pharmacist was unavailable. All involved pharmacists were successfully Personal Aseptic Technique Test (PATT) tested and employee training checklists were reviewed to ensure that all areas of sterile compounding training had been successfully completed. Lastly, a pharmacist swim lane document containing all steps in the process was developed to ensure consistency among staff members and these steps are mimicked in an EHR based checklist for ease of use, Figure 1.
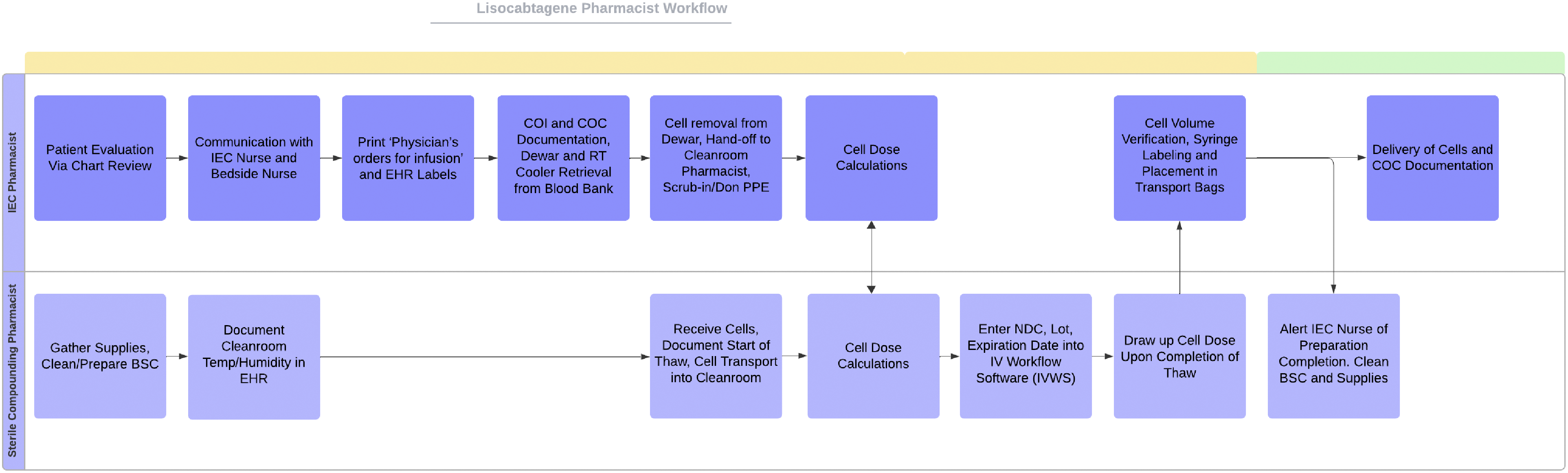
Swimlane diagram of pharmacist workflow.

Lisocabtagene program implementation timeline.
In addition to internal personnel training verification, FDA required REMS program training was also completed. REMS programs are a required component of the FDA approval for therapies that pose serious safety concerns. 15 They are designed to promote the safe use of medications by preventing, monitoring, and managing risks via education for those involved in the drug use process. In the context of lisocabtagene specifically, the REMS program stipulates, in part, that all “all relevant staff involved in the prescribing, dispensing, or administration of lisocabtagene are trained on the REMS requirements and must successfully complete the Knowledge Assessment and submit it to the REMS Program”. 16 While traditionally BMT pharmacists are REMS certified for all CART products, non-BMT pharmacists and pharmacy technicians need to enroll and gain certification if their responsibilities expand to include preparation of lisocabtagene. The three core pharmacists involved in program creation are responsible for lisocabtagene preparation; new pharmacist training time varies due to the inconsistent frequency of lisocabtagene preparation. Each pharmacist trained in this process must first complete the REMS required training and knowledge assessment and provide confirmation of completion which is documented. Pharmacists are also provided institution specific educational materials to review and finally, must shadow the core pharmacists during an actual dose preparation at least twice before being signed-off.
As described, training was validated with multiple trial runs with simulated patients. A demonstration kit was provided by the manufacturer that allowed for practice with empty vials and the packaging cassettes that encase the vials. Obtaining this demonstration kit provided staff with a means to anticipate appropriate handling. For example, the simple act of allowing sanitized, gloved hands to dry once working in a biological safety cabinet was a helpful consideration as the task of holding the long, thin vial while performing the aseptic manipulation is atypical in nature. The trial runs also allowed staff to don appropriate personnel protective equipment, handle Dewars on multiple occasions, and use cryogenic gloves. Lisocabtagene maraleucel as per the manufacturer's description is supplied in vials; the CD8 and CD4 components are packaged separately. However, the typical vial shape that most pharmacy teams are used to working with in practice is vastly different from the vials used for packaging lisocabtagene maraleucel. The vials are thin, 3-inch-long cylinders with two tubing lines attached to one end with a septum at the other. One line contains a filter which must be cut in a specific manner before the aseptic manipulation can commence. This mechanism then allows for the cellular product to be extracted using the negative pressure technique, where no air is added to the vial. Another interesting aspect is that the vial septum can only be punctured once and re-entry into the vial is not allowed. In addition to using the demonstration kits to prepare for cell extraction, laminated visual aids of the manipulation from the package insert were made available for use in the negative pressure room. This aseptic manipulation is a high stakes situation considering the patient specific nature of the product and the approximate cost of $480,000 per treatment based on WAC. 17
Conclusion
There is a need for regulatory agencies to provide consensus standards for cellular therapy product handling and preparation. If a cellular product is deemed anti-neoplastic after performing an assessment of risk and is classified as a hazardous drug, aseptic manipulation should be performed by the pharmacy department. Pharmacy teams should be performing these aseptic manipulations to be in line with USP <800 > and to help protect healthcare personnel that do not have access to engineering controls. As described by Marzal-Alfaro et al., pharmacists are integral to the successful management and safety of CART therapy programs and serve an important role within a multidisciplinary team. 18 Overall, the process took seven months; from the formulary addition request to treating our first lisocabtagene patient and completing a post-infusion analysis Figure 2. While our process was initially developed for lisocabtagene, it created a foundation on which we can onboard newly approved cellular or gene therapies that require manipulation. So far, the other medications on formulary at our institution have not required the same level of complexity in compounding. For example, etranacogene dezaparvovec-drlb, a gene therapy indicated for the treatment of hemophilia B, is stored under refrigeration (2–8°C), requires manipulation in a BSC and is supplied in kits containing 10 to 48 single use vials, the contents of which are added to 0.9% Sodium Chloride bags similar to other medications. 19 However, cellular therapies that are currently in the pipeline will require storage in vapor phase liquid nitrogen and extraction of cells from a thawed bag, in a BSC to achieve a patient specific dose like lisocabtagene.
At our institution, errors related to all aspects of the medication use process are documented in an internal electronic system; a search of that system revealed reports related to cellular therapies were associated solely with cellular therapy adverse reactions (i.e., cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome). There were no reported errors related to the thawing and compounding of cellular therapy products prior to or 4 months after program implementation; of the ten patients treated with lisocabtagene thus far, all doses were administered within the 2-h timeframe from thaw start to administration.
One challenge to implementation was the FACT requirement that humidity in the negative pressure room have an upper and lower range for monitoring purposes. Previously our pharmacy department SOPs stated that the humidity of the IV room suite would be monitored but did not specify an acceptable range nor a plan for when the room(s) were out of range. This required revisions to not only our departmental policy but also creation of a documented plan for out-of-range scenarios that would work across all of our IV room suites in the hospitals and infusions centers.
Regarding implementation of a similar program at other institutions there can be limitations in terms of generalizability, for example, not all institutions have an on-site negative pressure room where hazardous drugs are compounded. Due to the short expiration time after the start of thaw of most cellular products, utilizing an off-site IV room may not be feasible accounting for thaw time, preparation, and transport to the site of administration. Additionally, the lack of an investigational drug service with product experience prior to commercial availability which can inform new therapy onboarding, may slow down implementation. After implementation an oncology pharmacist listserv inquiry regarding pharmacist labeling and/or compounding of CART products revealed that most health systems rely on the blood bank or stem cell processing laboratory to manipulate and label all CART products including lisocabtagene. Of ten respondents, one institution manipulated lisocabtagene in the pharmacy, and five of ten utilized a pharmacist to label CART products.
The FACT accreditation process is an essential component of cellular therapy programs and successful process validation requires a multidisciplinary team of experts. Our experience described here demonstrates the successful implementation of providing cellular therapy products to patients at a multisite academic medical center utilizing the inpatient pharmacy department for aseptic manipulation of cells.
Footnotes
Author contributions
JGM drafted portions of the manuscript and compiled the final version. KM drafted portions of the manuscript. LG drafted portions of the manuscript. BD reviewed the final manuscript and provided expert opinion. JJ drafted sections of the manuscript. All authors reviewed and approved the final version of the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.