
Abstract
Background
Systemic mastocytosis (SM) is a rare and potentially severe hematologic disorder characterized by the clonal proliferation of mast cells (MCs) into various organs. The clinical manifestations of advanced SM are caused by the uncontrolled release of cytokines and vasoactive amines from MC and disease-induced organ dysfunction. Patients with SM typically present with symptoms such as flushing, pruritus, diarrhea, and headaches, but outcomes following active treatment have not been well characterized. In this study, the clinical characteristics, treatment patterns, and natural history of an SM patient cohort diagnosed and treated within a community hematology network in the United States is described.
Methods
We identified 105 patients who were diagnosed and managed in one of 19 community hematology clinics up to an index date of 1 October 2022. Data collection included patient and disease characteristics, baseline biochemistry and hematology, SM diagnostic criteria being met, biomarkers tested, CD2 and/or CD25 expression in MCs as well as serum tryptase levels at presentation. Data abstraction also included supportive care drugs and anticancer therapy used, treatment response, reason for discontinuation, and overall survival by disease subtype.
Results
A total of 105 SM patients were identified who met the inclusion criteria. The specific SM subtypes were indolent (47.6%), aggressive (9.5%), SM with an associated hematological neoplasm (19.0%), MC leukemia (1.9%), and subtype not documented (21.9%). Regardless of subtype, approximately 62% of patients did not receive SM-directed active therapy. Only 26% of patients with indolent systemic mastocytosis (ISM) received treatment compared to 65.6% with advanced subtypes. Relative to ISM cohort, the relative risk of death in patients with the advanced SM subtypes was approximately 15 times greater (hazard ratio = 15.0; 95% confidence interval: 3.3 to 66.5).
Conclusions
SM patients present with multiple underlying symptoms, within various disease subtypes that are difficult to diagnose in a timely manner. As a result, many patients do not receive active drug therapy for their disease. Therefore, greater disease awareness is required as well as new tools for earlier disease detection.
Introduction
Systemic mastocytosis (SM) is a rare hematological disorder that affects approximately 1 in every 10,000 to 20,000 people worldwide. 1 It can occur in people of any age, but is most commonly diagnosed in adults over the age of 50.1,2 The disorder is characterized by the accumulation and activation of mast cells (MCs) in various tissues and organs of the body, including the skin, bone marrow, liver, spleen, and gastrointestinal tract.3,4 MCs are a type of white blood cell (WBC) that play a crucial role in the immune system by releasing histamine and other chemicals in response to allergens and other stimuli. In SM, the excessive and uncontrolled activation of MCs can lead to a wide range of symptoms, including skin lesions, flushing, itching, abdominal pain, diarrhea, nausea, vomiting, headaches, fatigue, bone pain, and muscle weakness.2,3
The severity and frequency of these symptoms can vary widely among patients based on disease subtype. SM is subdivided into five different subtypes: indolent systemic mastocytosis (ISM), smoldering systemic mastocytosis (SSM), aggressive systemic mastocytosis (ASM), SM with an associated hematological neoplasm (SM-AHN), and mast cell leukemia (MCL).4,5 Further segmentation includes non-advanced SM, consisting of ISM and SSM, and advanced (AdvSM), comprising ASM, SM-AHN, and MCL. Patients diagnosed with non-advanced SM experience symptoms such as flushing, pruritus, diarrhea, and headache.5,6 With the more advanced disease subtypes—ASM, SM-AHN, and MCL—patients have a poor prognosis and shorter overall survival (OS).5,6
SM is usually caused by mutations in the KIT gene, which provides instructions for making the KIT receptor that is involved in the regulation of MC growth and survival.5,7 However, the exact cause of the disorder remains unknown, and additional genetic and environmental factors may also play a role.1,2 There is currently no cure for the majority of SM patients, and treatment is typically directed at symptom palliation and preventing complications. This may include supportive care agents such as antihistamines, corticosteroids, MC stabilizers, as well as avoiding triggers through lifestyle modifications.5–7 Historically, there have been no treatments that target the underlying cause of the disease. In the advanced subtypes, ASM, SM-AHN, and MCL, more aggressive treatments are often offered to patients. These include agents such as interferon, hydroxyurea, midostaurin, imatinib, cytarabine, as well as bone marrow transplantation, which is used with a curative intent.5–7
As described earlier, SM is usually caused by mutations in the KIT gene. Small molecule tyrosine kinase inhibitors targeting key KIT mutations such as D816 V have changed the natural history of patients with AdvSM and have improved symptom control.7,8 Such agents include imatinib, midostaurin and avapritinib. However, only the latter two have received regulatory approval in the United States and Europe for patients with AdvSM. Midostaurin was approved based on the results of an open label phase II trial. In that study, 116 patients, 89 of which had disease-related organ damage, received midostaurin at a dose of 100 mg twice daily. The overall response rate was 60%, with major response being seen in 45% of patients. 9 However, complete responses were not observed. The investigators reported that 56% of patients required dose reductions because of toxicity. However, reescalation to the original starting dose was feasible in 32% of those patients. 9
Avapritinib, a small molecule that targets the KIT and PDGFRA A-loop mutants including D816 V was the second agent to receive approval in the United States and Europe for AdvSM. 8 Clinical trials (e.g. Explorer, Pathfinder) demonstrated that avapritinib generated high response rates, including complete responses in AdvSM.10,11 Patients also had symptomatic disease improvement and a reduction in their bone marrow MC burden. Most importantly, approximately 33% of patients had molecular complete remission and KIT D816 V was no longer detectable in the marrow.10,11 The drug was well tolerated and there were no patients coming off study because of toxicity. KIT D816 V mutations are also the underlying cause of most cases of indolent SM.7,8 Recently, a placebo-controlled randomized trial demonstrated symptomatic and objective benefits as well as an improvement in quality of life (QOL) with avapritinib in patients with ISM. 12 There were no new safety signals, adverse events were mostly grade 1, and there were no treatment-related discontinuations. 12 The Food and Drug Administration of the United States approved avapritinib for ISM in May 2023.
The Quality Cancer Care Alliance (QCCA) is a network of 19 community oncology/hematology practices across the United States. SM is a rare hematological disorder, which has not been well characterized outside of the clinical trial setting. In this study, the clinical characteristics, treatment patterns, and natural history of SM patients diagnosed and treated within the real-world QCCA network are described.
Methods
Patients
This was a retrospective observational study consisting of patients with SM who presented to a community hematology practice for treatment. The time interval for patient management was between 1 July 2002 to 1 October 2022. All patients received treatment in one of 19 community hematology practices that are part of the QCCA network. To be entered into the study, patients were ≥ 18 years of age and diagnosed with SM regardless of subtype.
Data collection
Data was collected from the electronic medical records (EMRs) of QCCA clinical practice sites. It consisted of patient demographics, disease characteristics and subtype, clinical signs and symptoms at presentation, duration of SM symptoms prior to the diagnosis, patient performance status at presentation and existing comorbidities as assessed by the Charlson comorbidity index. 13 Additional information collected included the number of major and minor SM diagnostic criteria met, MC morphology, biomarkers tested, CD2 and/or CD25 expression in MCs as well as serum tryptase levels at presentation.
From date of diagnosis until the end of the patient record, data were collected on hemoglobin (Hb), WBCs, absolute neutrophil count, platelet counts, serum tryptase levels, active drug therapies delivered as initial therapy, and the use of supportive care agents. Data abstraction also included the starting dose of the drug therapy used to target SM, disease response to this therapy, reason for discontinuation, and OS by disease subtype. All information was collected via a standardized electronic data collection form. Specifically, for patients that met the eligibility criteria, the data was centrally collected by a team of curators using the electronic data collection form. All curated data was deposited into a secure database, for the subsequent statistical analysis. The data collection form is available upon request to the corresponding author.
Sample size and statistical considerations
The main endpoints in this descriptive, observational study were related to disease-related symptoms at presentation and treatment delivered by disease subtype. Patients were subdivided into three SM cohorts: ISM, AdvSM, and SM with an undocumented subtype (SM-UD). Exploratory endpoints consisted of OS and response to therapy by subtype. A formal sample size calculation was not performed and data on all eligible patients treated up to 1 October 2022 were collected.
Patient demographics, clinical features, and frequency of disease-related symptoms were presented descriptively as means, medians, or proportions, with standard deviations and 95% confidence intervals (CIs). In the exploratory analysis, OS by SM subtype and cohort was assessed as of the index date (1 October 2022) using Kaplan–Meier (KM) methods and hazard ratios (HR) were calculated using univariate Cox regression analysis. All statistical analyses were performed using Stata, release 16.0 (Stata Corp., USA).
Results
A total of 105 SM patients were identified within the QCCA real-world database. The specific SM subtype diagnoses of these patients were ISM (47.6%), aggressive SM (9.5%), SM-AHN (19.0%), MCL (1.9%), and undocumented (SM-UD) (21.9%). The distribution by subtype for the current analysis consisted of 50 (47.6%), 32 (30.5%), and 23 (21.9%) patients in the ISM, AdvSM, and SM-UD cohorts respectively. It is important to note that among the ISM cohort, there were two patients who were diagnosed with the smoldering SM subtype (SSM). However, with only two patients, it would have been untenable to have an SSM cohort for analysis. As a result, the two patients were merged into the undocumented SM cohort for the survival analysis.
Patients with AdvSM tended to be older, male gender have more underlying comorbidities, more likely to have spleen or lymph node enlargement and less likely to have cutaneous mastocytosis prior to diagnosis (Table 1). In contrast, there was reasonable balance between cohorts in mean patient body mass index, race, performance status, multifocal dense infiltrates of MCs, and the proportion of patients meeting the major diagnostic criteria for SM, established by the World Health Organization (WHO). 13 However, there was variability between groups in the proportion of patients with serum tryptase level >20 ng/mL. 50.0%, 53.1%, and 34.8% of patients in the ISM, AdvSM, and SM-UD cohorts had a KIT point mutation at codon 816 (Table 1); of note, there was limited use of high specificity testing for KIT mutations among patients tested for KIT mutations, likely resulting in a high number of false negative results.
Demographic and clinical characteristics of patients by SM subtype at diagnosis.
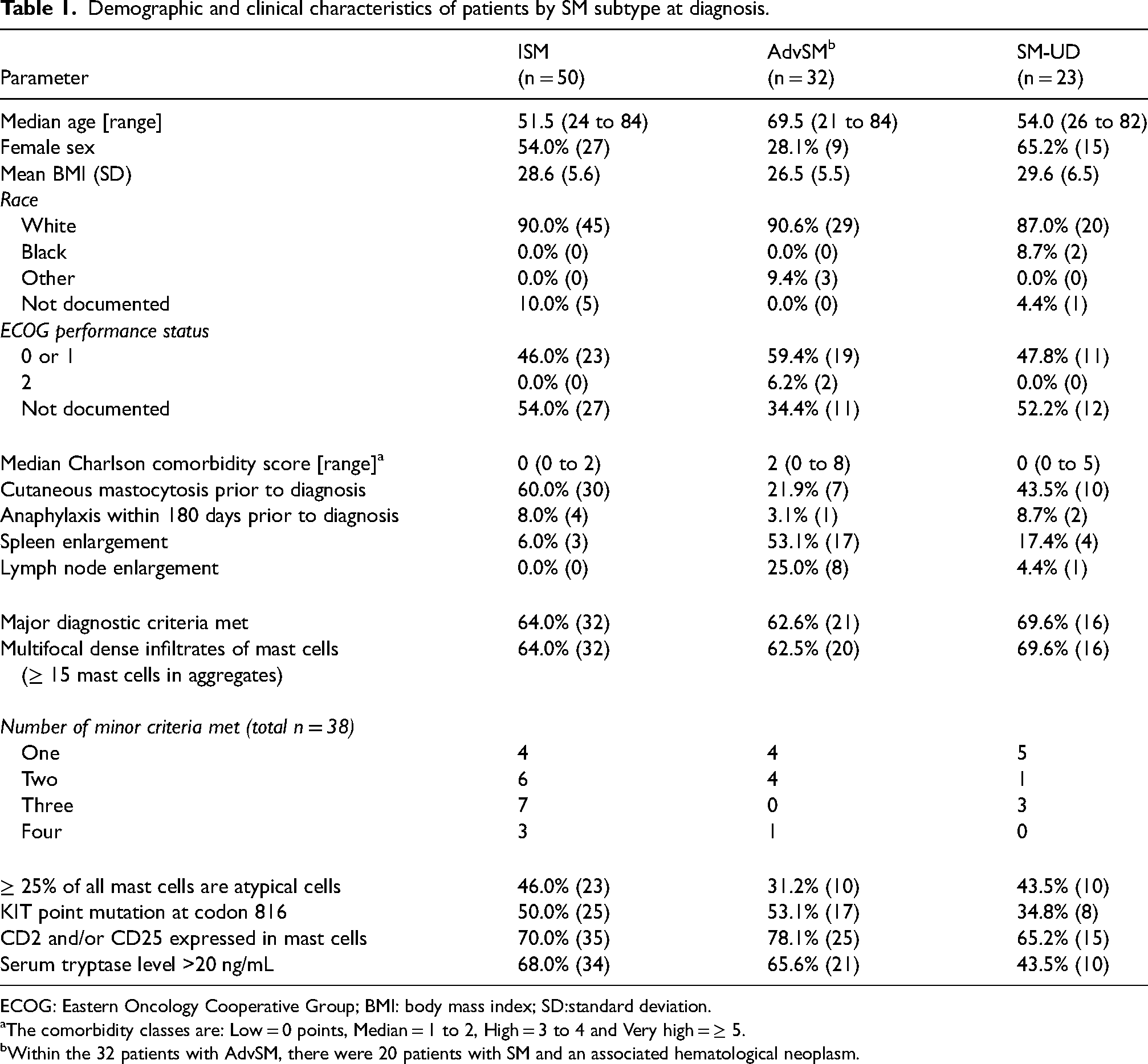
ECOG: Eastern Oncology Cooperative Group; BMI: body mass index; SD:standard deviation.
The comorbidity classes are: Low = 0 points, Median = 1 to 2, High = 3 to 4 and Very high = ≥ 5.
Within the 32 patients with AdvSM, there were 20 patients with SM and an associated hematological neoplasm.
The analysis was continued with an evaluation of patient symptoms and laboratory parameters within 30 days of diagnosis. Symptoms such as diarrhea, fatigue, hypertension, cutaneous reactions, and weight loss were common among all three cohorts, but appeared to be most prevalent in patients with AdvSM (Table 2). However, renal function, standard biochemistry, and hematological parameters (e.g., Hb, platelets, WBCs, and neutrophils) appeared to be within normal limits for most patients. Consistent with a diagnosis of SM, mean serum tryptase levels were elevated beyond normal for patients in all three cohorts (Table 2). In at least 30% of patients within each cohort, the bone marrow was also infiltrated by abnormal MCs that were spindle shaped or were atypical (Table 2). The observed morphological and functional changes in MCs are relevant because they contribute to the symptoms and clinical features of SM. CD25 is a biomarker for MC activation and proliferation in SM. CD2 positivity on MCs is associated with the advanced variants of the disease.3,4 Across all three patient cohorts, at least 65% of patients were positive for CD25. CD2 positivity varied between 15.6% and 44.0% across the three patient cohorts. With respect to the testing of mutations prior to the start of initial therapy, the only one that was tested in at least 50% of patients was KIT D816 V (Table 2).
Patient symptoms and laboratory parameters at diagnosis.
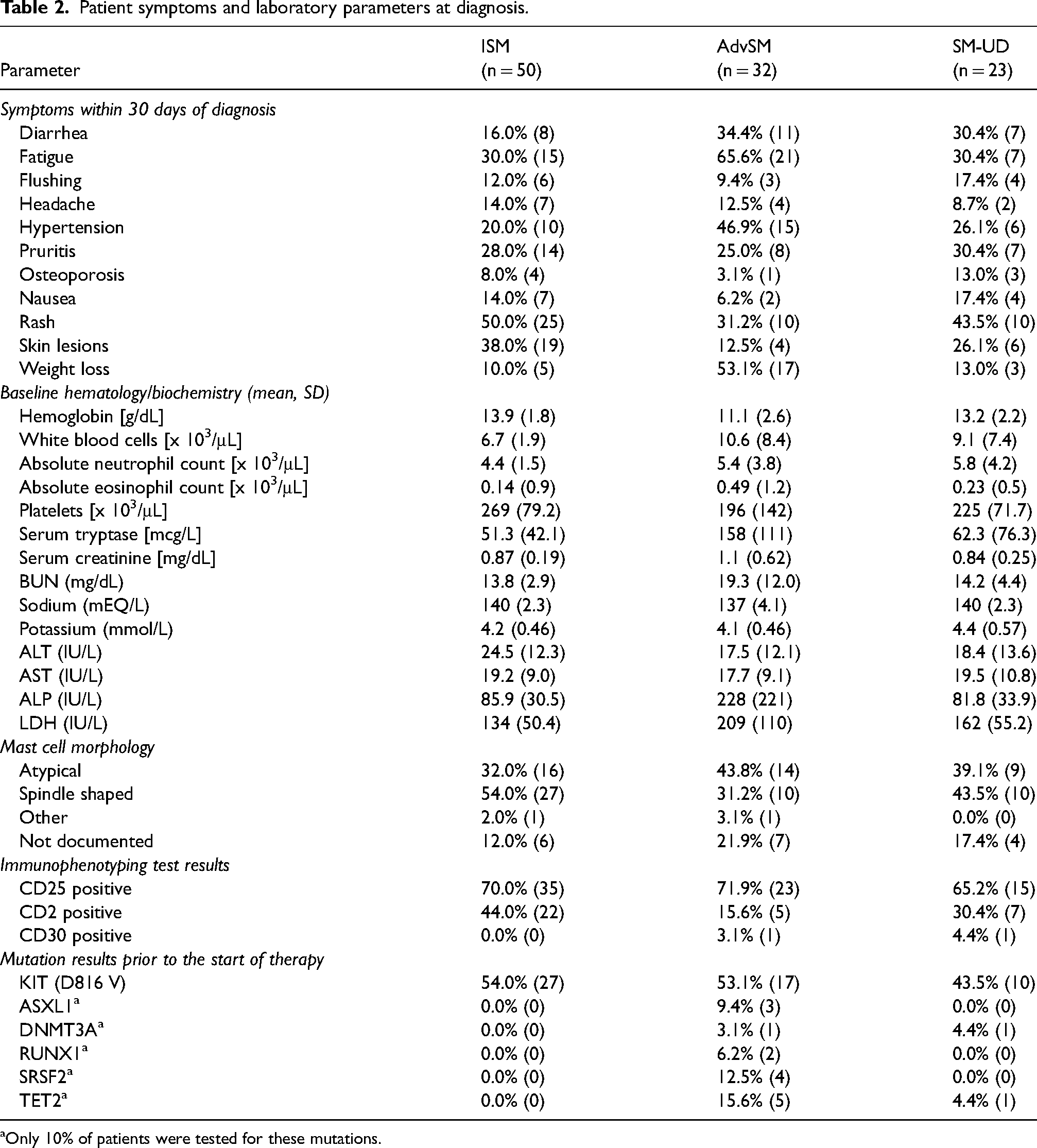
Only 10% of patients were tested for these mutations.
Primary therapy intended to arrest disease progression and supportive care drug use for symptom relief for newly diagnosed SM patients was evaluated. At presentation, 62.0%, 59.4%, and 60.9% of patients in the ISM, AdvSM, and SM-UD cohorts were receiving supportive care consisting of antihistamines, corticosteroids, cromolyn sodium, and leukotriene receptor antagonists. When active drug therapy for disease control was assessed, there was no documented treatment in approximately 74% of patients with ISM or in the cohort with undocumented SM subtype (Table 3). It is important to note that over the study period, there were no approved drug therapies for ISM. In contrast, 65.6% of patients with AdvSM received multiple initial therapies such as avapritinib, azacitidine, cladribine, hydroxyurea, and imatinib. The most common agent used was midostaurin, which was prescribed in 25% of patients (Table 3). In patients who received active drug therapy, the median duration was 8.4, 5.5, and 12.2 months in the ISM, AdvSM, and SM-UD cohorts, respectively (Table 3). Most patients in each cohort were able to achieve at least disease stabilization. Indeed, 4 of 21 (19.0%) patients in the AdvSM cohort who received active treatment achieved a complete or partial response.
Primary SM therapy following diagnosis and associated clinical outcomes.

IQR: inter quartile range; HR: hazard ratio.
32 patients had a practice interaction on or after 10/01/2022.
The two patients with smoldering SM were grouped with the not documented cohort for the survival analysis.
A hazard ratio of > 1.0 suggests an increased risk of death and < 1.0 a reduced risk of death.
The final clinical outcome assessed by patient cohort and disease subtype was OS. As of the index date (1 October 2022), the mortality rate was 4.0%, 40.6%, and 13.0% in the ISM, AdvSM, and SM-UD cohorts, respectively (Table 3). Using the ISM cohort as a reference, the HR for risk of death for the AdvSM and SM-UD cohorts was 15.0 (95%CI: 3.3 to 33.5) and 3.0 (95%CI: 0.5 to 18.2), respectively. The KM OS curves confirmed that patients with AdvSM had the poorest survival outcomes (Figure 1). Within the 32 patients in the AdvSM cohort, there were 20 patients with SM-AHN. KM OS curves were regenerated with the 20 SM-AHN patients as a stand-alone subgroup. The findings suggested that the SM-AHN subtype was associated with the poorest prognosis, with the median OS being approximately 5 years from diagnosis (Figure 2). When the risk of death in patients with SM-AHN was compared to those with ASM, the HR was 3.89 (95%CI: 0.85 to 17.9).
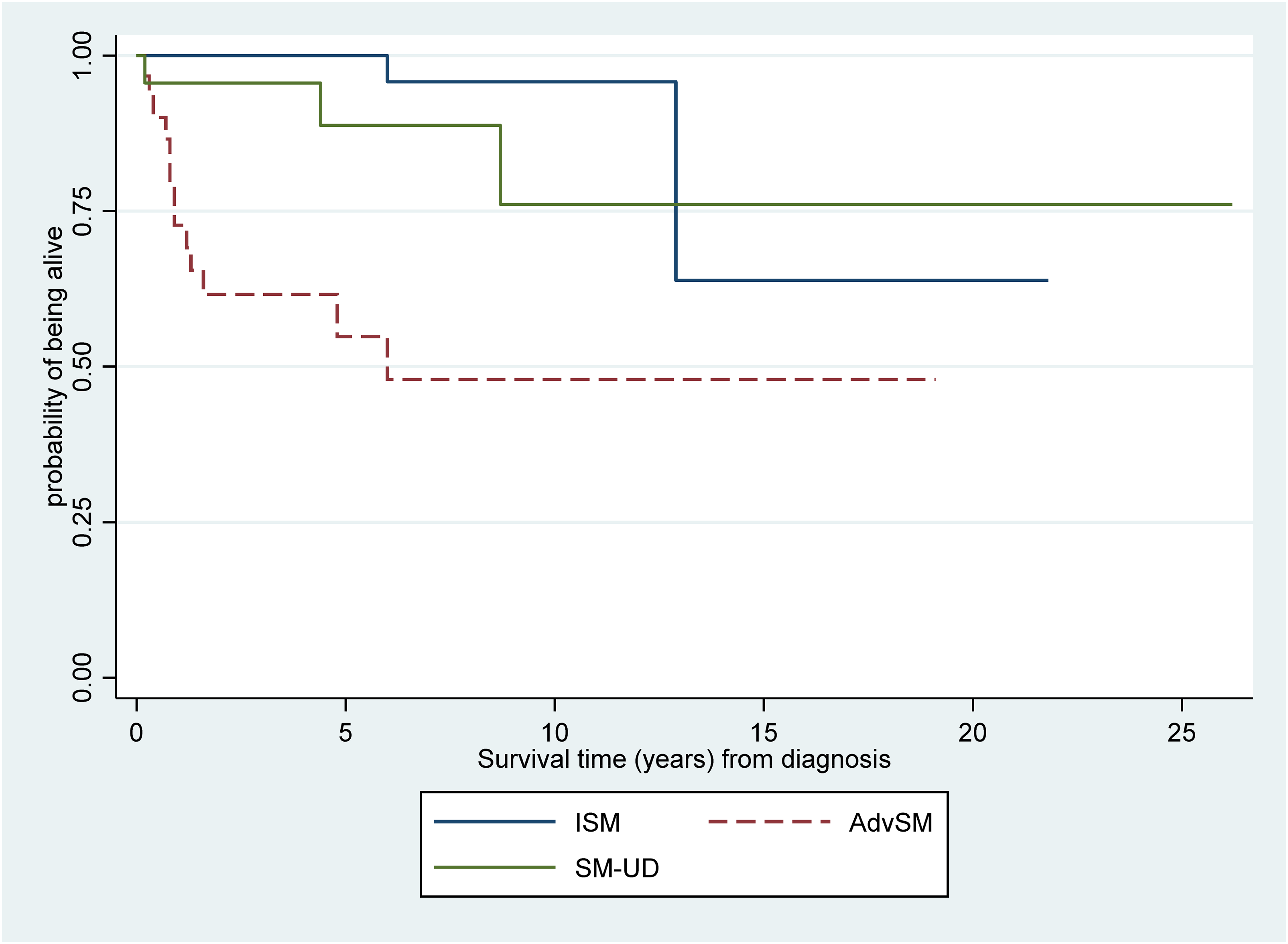
Overall survival by SM cohort.
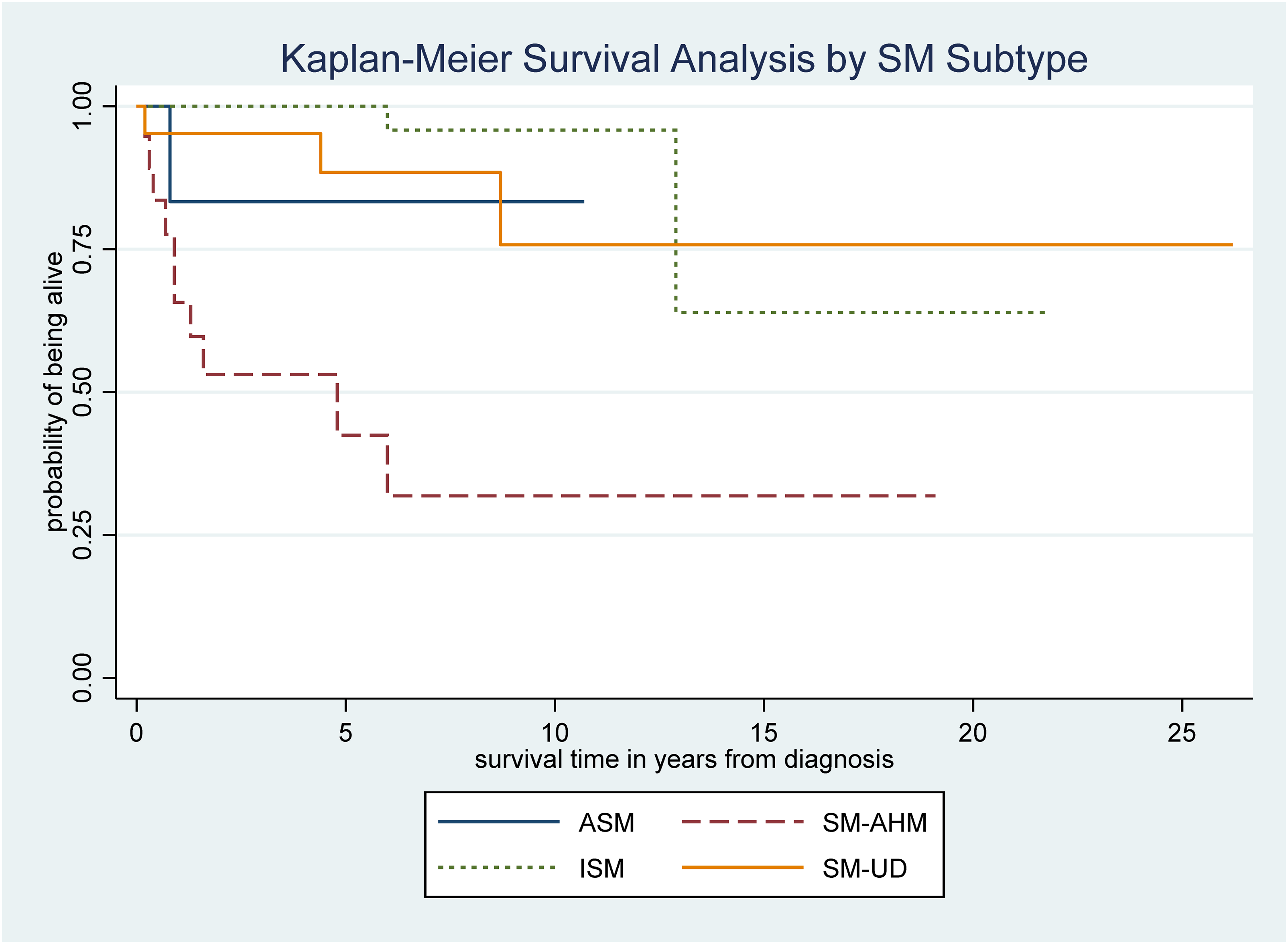
Overall survival by SM subtype.
Discussion
SM is a rare and potentially severe hematologic disorder that has not been well described in the context of community cancer care. To that end, we conducted a descriptive evaluation of patients treated at community hematology practice sites across the United States. One of our primary findings was that community-based hematologists see SM patients of all subtypes, but the most common appears to be ISM. Patients also appear to present with the hallmark immune symptoms of SM such as flushing, pruritis, rash, and skin lesions. Indeed, all patients with ISM and AdvSM had at least one of these symptoms. Despite this, most patients at presentation appeared to have hematology and biochemistry values within normal limits. However consistent with a diagnosis of SM, patients in all three cohorts had significantly elevated serum tryptase levels at presentation. Indeed, serum tryptase levels >25 ng/ml were identified as a statistically significant predictor of an SM diagnosis.14,15 In addition, approximately 65% of patients in all cohorts had met the major diagnostic criteria proposed by the WHO. 15
Given the fact that SM is a rare disease, a timely diagnosis can be challenging. In our sample of 105 SM patients, 21.9% did not have a documented disease subtype, suggesting difficultly in the accurate diagnosis of this rare disorder. Given the need for a rapid and accurate diagnosis of SM in the community setting, we have developed and externally validate a diagnostic algorithm for use by the practicing clinician. 16 The clinical utility of this tool will be for the earlier detection of SM patients presenting to primary or secondary care physicians.
Following diagnosis, our study evaluated the use of active interventions in the management of SM patients. Only 26% of patients with ISM received active drug therapy compared to 65.6% with AdvSM. One probable reason why fewer patients with ISM received active therapy could be due to limited clinical data indicating symptomatic benefit in such patients. However, the recent placebo-controlled trial that evaluated avapritinib in ISM patients reported clinical benefit and an improvement in QOL. 12 As the clinical data from this trial becomes more widely disseminated, we anticipate that more patients with ISM will be receiving active therapy that is evidence based.
Our study evaluated patient outcomes in terms of disease response and OS. Most patients, regardless of cohort, who received active therapy achieved at least stable disease. The data also revealed that patients with ISM had the longest OS, with the median not having been reached. The findings also confirmed that patients with AdvSM had a worse overall prognosis. Relative to the ISM cohort, the risk of death in AdvSM patients was approximately 15 times greater (HR = 15.0; 95%CI: 3.3 to 66.5). When the risk of death in patients with SM-AHN was compared to those with aggressive SM only, the HR was 3.89 (95%CI: 0.85 to 17.9), with the median OS being less than 5 years from diagnosis, further demonstrating the severity of the SM-AHN subtype and underscoring the need for active therapy that will prolong OS.
To our knowledge, the current study is one of the largest to evaluate the natural history of SM patients with various disease subtypes managed in a real-world community setting. Notwithstanding, there are important limitations that need to be acknowledged. Given that SM is a rare disease, the sample size was small. There was undocumented data for several important parameters such as patient performance status, response to therapy, and date of death. The sample size was limited to the community setting of the United States. This limits the generalizability of the findings to patients treated in academic centers or in other countries. For the survival analysis, all patients who were alive or lost to followed as of 1 October 2022 were censored. However, because of the long time period of patient management (1 July 2002 to 1 October 2022), we acknowledge unequal follow-up for patients within each SM cohort and this may have biased the survival analysis.
Conclusions
In this retrospective evaluation of SM patients managed in a real-world community hematology setting, it was determined that patients present with multiple underlying symptoms, among the various disease subtypes that are often difficult to diagnose in a timely manner. As a result, many patients do not receive active drug therapy for their disease, even in those with more advanced subtypes and a poorer overall prognosis.
Footnotes
Author contributions
GD, HN, AP, and TG2 conceived of the study and managed the overall direction, and planning of the study. GD, HN, DP, KM, and TG1 provided support to the design and implementation of the data collection. HN and AP were responsible for administrative, technical, or material support. GD and HN prepared the tables and figures. DP, KM, TG1, and TG2 provided clinical input in the interpretation of the data and critical review of the manuscript. GD conducted the statistical analysis and wrote the initial manuscript, and all authors were involved in interpreting the results, editing and critically reviewing the manuscript.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: DP, KM, and TG are employees and shareholders of the sponsor (Blueprint Medicines Corporation).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this study was provided by Blueprint Medicines Corporation.
Statement of ethics
The study was not initiated until the protocol was reviewed and approved by a centralized institutional review board (IRB). The IRB determined that the study was exempt from oversight due to the retrospective nature of the data collection.