
Abstract
Background
Despite robust evidence and international guidelines, to support routine pharmacogenetic (PGx) testing, integration in practice has been limited. This study explored clinicians’ views and experiences of pre-treatment DPYD and UGT1A1 gene testing and barriers to and enablers of routine clinical implementation.
Methods
A study-specific 17-question survey was emailed (01 February–12 April 2022) to clinicians from the Medical Oncology Group of Australia (MOGA), the Clinical Oncology Society of Australia (COSA) and International Society of Oncology Pharmacy Practitioners (ISOPP). Data were analysed and reported using descriptive statistics.
Results
Responses were collected from 156 clinicians (78% medical oncologists, 22% pharmacists). Median response rate of 8% (ranged from 6% to 24%) across all organisations. Only 21% routinely test for DPYD and 1% for UGT1A1. For patients undergoing curative/palliative intent treatments, clinicians reported intent to implement genotype-guided dosing by reducing FP dose for DPYD intermediate metabolisers (79%/94%), avoiding FP for DPYD poor metabolisers (68%/90%), and reducing irinotecan dose for UGT1A1 poor metabolisers (84%, palliative setting only). Barriers to implementation included: lack of financial reimbursements (82%) and perceived lengthy test turnaround time (76%). Most Clinicians identified a dedicated program coordinator, i.e., PGx pharmacist (74%) and availability of resources for education/training (74%) as enablers to implementation.
Conclusion
PGx testing is not routinely practised despite robust evidence for its impact on clinical decision making in curative and palliative settings. Research data, education and implementation studies may overcome clinicians’ hesitancy to follow guidelines, especially for curative intent treatments, and may overcome other identified barriers to routine clinical implementation.
Introduction
Fluoropyrimidines (5-Fluorouracil and capecitabine, FP) and irinotecan are commonly used chemotherapies, in gastrointestinal cancers and other solid tumours. Patients carrying specific DPYD variant allele(s) or homozygous for the UGT1A1*28 are at increased risk of serious and life-threatening toxicities from FP and irinotecan, respectively.1–3
There is a growing body of evidence for pharmacogenetic (PGx) guided prescribing for these drugs, with major guidelines published by the Dutch Pharmacogenetics Working Group (DPWG) and Clinical Pharmacogenetics Implementation Consortium (CPIC).1–3 The European Medicines Agency (EMA) and the United Kingdom government have recommended PGx testing prior to FP chemotherapy. 4 Additionally, the United States Food and Drug Administration (FDA) recommends dose reduction in patients who are homozygous for the UGT1A1*28 allele prior to irinotecan chemotherapy. In September 2022 the Australian Therapeutic Drug Administration (TGA) has updated the warning for FP, advising that patients with complete DPD deficiency should not be treated with FP and a reduced starting dose should be considered in patients with partial DPD deficiency. 5
Available evidence suggests that pre-treatment PGx-guided dosing in DPYD variant allele carriers is a promising strategy for the prevention of serious and potentially life-threatening FP-related toxicity without impacting on treatment outcomes.4,7,8 Large prospective multicentre studies from the Netherlands and United States (US), support the feasibility and/or cost-effectiveness of prospective DPYD genotyping for patients receiving FP across all tumour types.7–10 A soon to be reported trial will provide data in the Australian context (Australian New Zealand Clinical Trials Registry ANZCTR 12621000251820).
Similar to FP, international evidence from prospective studies demonstrates the importance of irinotecan dose adjustments especially in patients who are homozygous for the UGT1A1*28 allele.6,10 Despite the strong evidence and the known associations between DPYD/UGT1A1 specific variants and FP/irinotecan-associated toxicities, PGx testing and PGx-guided dosing has not been widely adopted, especially in Australia. There is upcoming PGx research with scope focusing on research priorities around PGx testing and quality use on medicines in Australian health care systems. The attitudes of oncology health professionals in regard to routine PGx testing are unknown. The study, representing the first survey of clinicians’ views and experiences of DPYD and/or UGT1A1 gene testing prior to FP/irinotecan chemotherapies, aimed to identify barriers and enablers to implementation.
Methods
Study development, sample selection and survey distribution
A study-specific 17-question survey was developed by a multidisciplinary expert panel including medical oncologists, oncology pharmacists, and researchers. The survey is composed of a series of quantitative questions that included multi-item or single-item scale(s) and one final open-ended question
The survey was first distributed and remained open for completion between February and April 2022. Email reminder was sent 2 weeks after the initial invitation and before closing date. A decision to click on the survey link by the participants and to complete/submit the survey was taken as informed consent. Participation was optional and there was no compensation offered for survey completion.
Survey analysis
Descriptive statistics were utilised to summarise survey responses. The results were reported as a proportion of respondents that answered each particular question, which may be lower than the overall number of respondents due to skipped/omitted responses. Results were analysed overall, by craft group (medical oncologist and pharmacist), and practice setting (Australia and International).
Results
Survey respondents
A total of 156 survey responses were initiated and partially completed via REDCap and 125 completed surveys were collected. Median response rate of 8% (ranged from 6%-24%) across all organisations. The majority of responses were from medical oncologists 78% (122/156) and Australian practitioners 82% (128/156). Respondents practised in varied clinical settings including metropolitan general hospitals 43% (67/156), metropolitan cancer hospitals 32% (50/156), regional general hospitals (22% (34/156), and regional cancer hospitals 13% (20/156)) (Table 1). The majority of responses were from public hospital-based clinicians 78% (122/156); 60% (94/156) had a greater than five years oncology experience; 26% (41/156) had more than 20 years’ experience, and 60% reported that they regularly treated cancer patients (Table 1).
Respondent demographics.
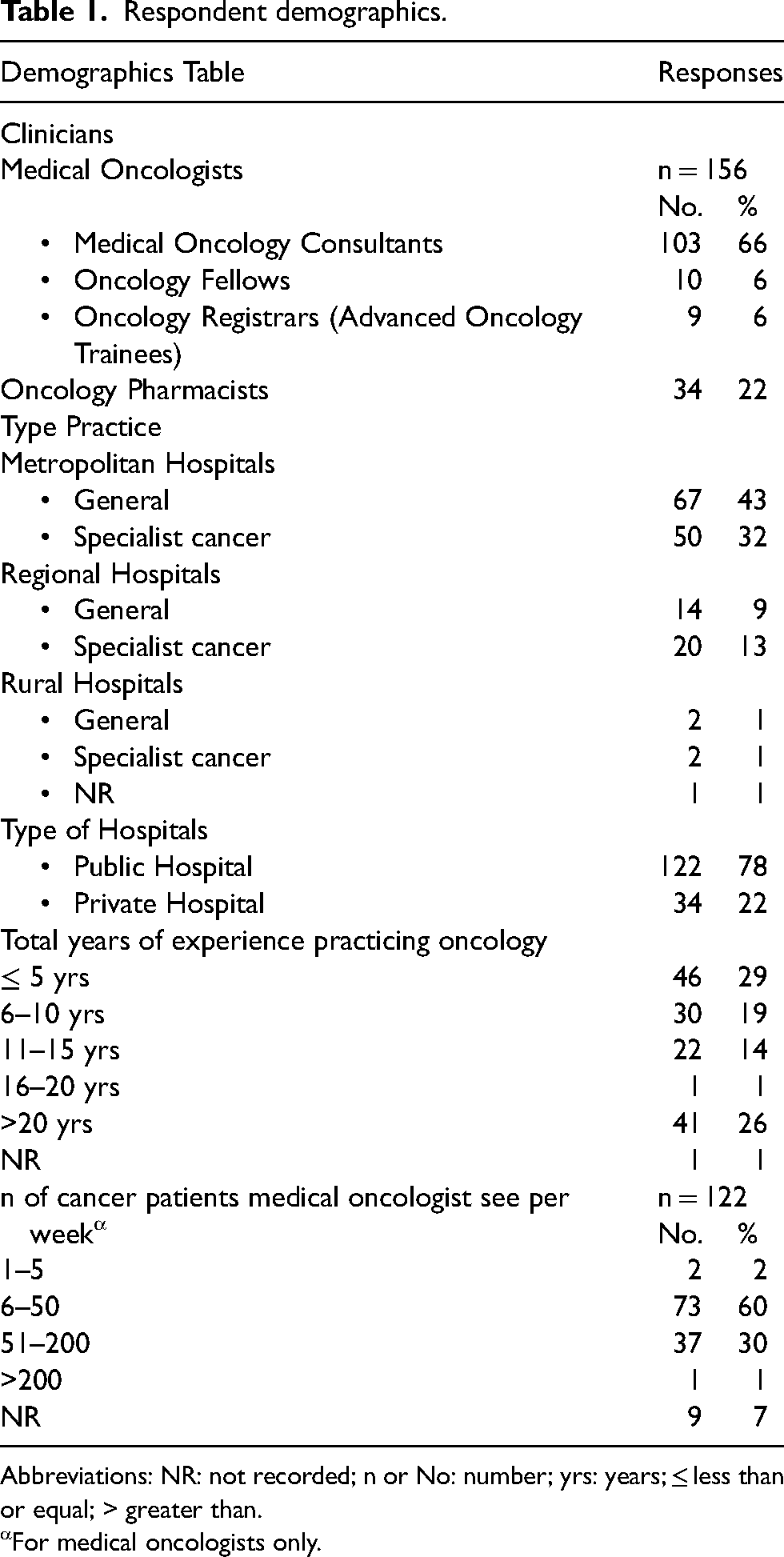
Abbreviations: NR: not recorded; n or No: number; yrs: years; ≤ less than or equal; > greater than.
For medical oncologists only.
DPYD and UGT1A1 gene testing before fluoropyrimidine and irinotecan chemotherapy
Only 21% (32/156) of clinicians reported testing for DPYD as part of routine care and 12% (18/156) as part of a clinical trial. Very few clinicians reported testing for UGT1A1 as part of routine care or a clinical trial (1% and 2%)
More clinicians rated very valuable or valuable in having DPYD testing before treatment with FP 77% (121/156) versus 53% (84/156) for UGT1A1 (prior to irinotecan), to help with treatment decision-making as part of usual care (Figure 1a).
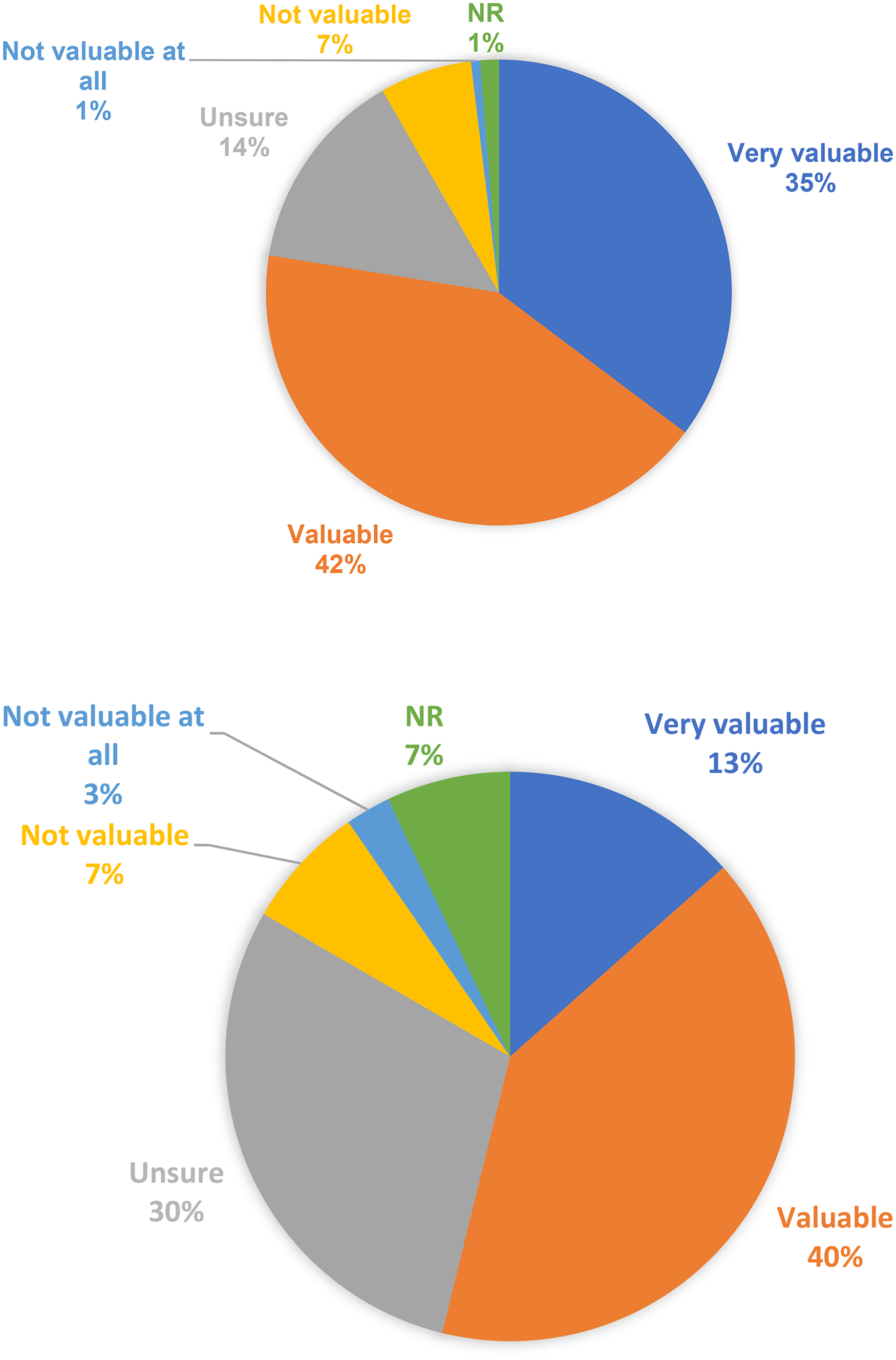
a: Clinician reported value of DPYD gene testing before commencement of fluoropyrimidines. Abbreviation: NR: not recorded. b: Clinician reported value of UGT1A1 gene testing before commencement of irinotecan. Abbreviation: NR: Not recorded.
Forty-six clinicians (30%) selected that they were unsure in having UGT1A1 gene test to help with irinotecan treatment decision making, compared to 14% (22/56) with regard to FP dosing (Figure 1b). Similar results were reported among medical oncologists and pharmacists, and among Australian and international practitioners.
Timelines for acceptable pre-treatment PGx screening
Sixty-three clinicians (40%) specified acceptable turn-around time for PGx screening of up to seven days business days in palliative settings, and up to five business days in curative settings 41% (64/156) (Figure 2).
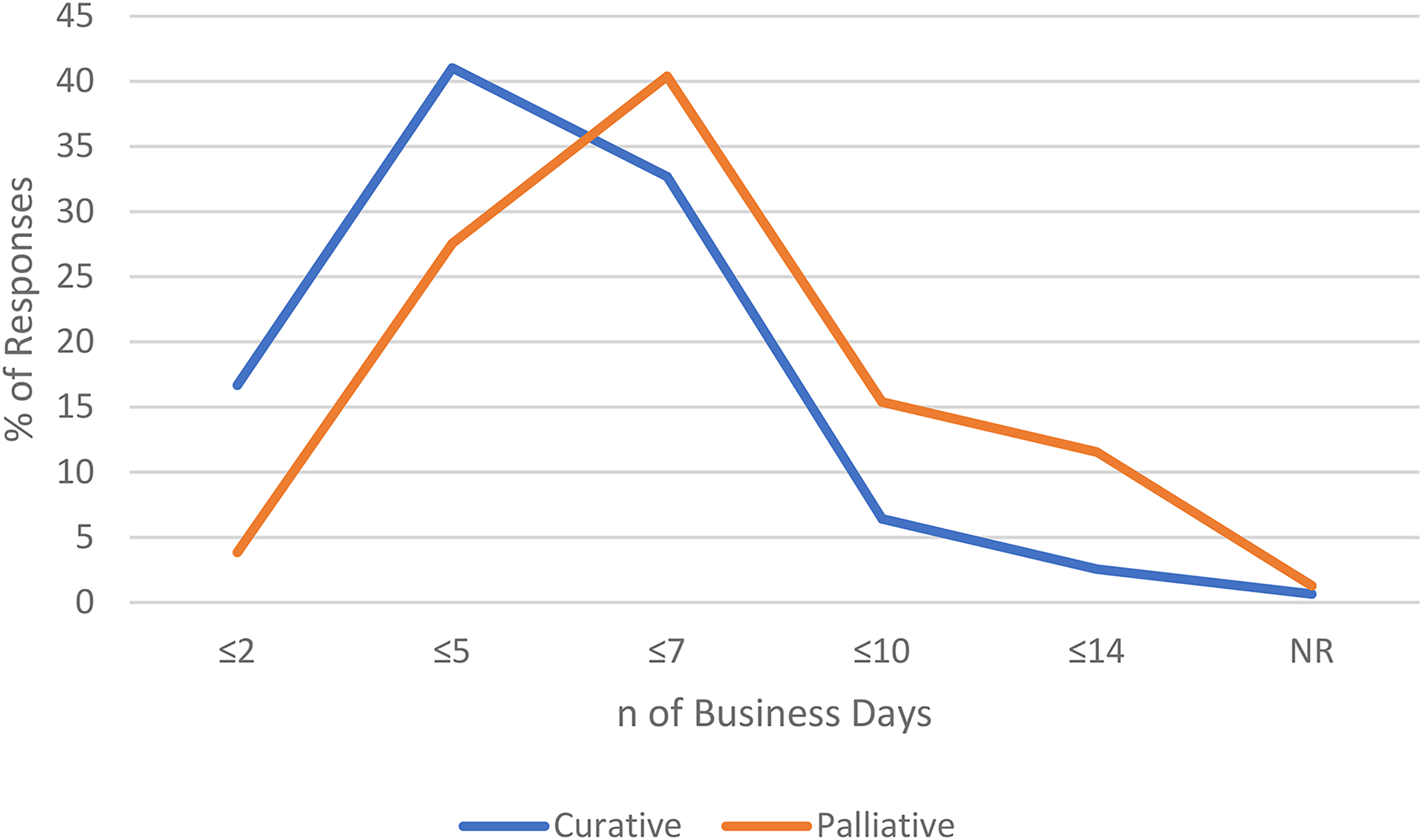
Clinician reported acceptable turnaround time for pharmacogenetic screening, according to treatment setting (curative and palliative). Abbreviations: n: number; % percentage; ≤ less or equal.
Impact of PGx on clinical decision making
When presented with case-based questions that would confer dose reduction or avoidance according to international guidelines from CPIC/DPWG, respondents reported varied clinical decision making, particularly in the curative intent setting. There was general agreement among the clinicians (selected definitely or probably) that FP doses should be reduced in the palliative treatment settings for patients who are DPYD intermediate metabolisers (IM) 94% (147/156) and to avoid FP in patients who are DPYD poor metabolisers (PM) 91% (141/156) as per the CPIC guidelines
Barriers and enablers for the implementation PGx services
The survey presented 12 potential barriers and enablers (Supplementary Material 1) asking respondents to rank their relevance and impact. The greatest barrier to routine implementation of PGx screening program identified by 82% (128/156) of respondents was the lack of government financial reimbursements, which in the Australian context refers to (Medical Benefit Scheme [MBS] funding). Other barriers included perceptions of lengthy PGx test turnaround time 76% (118/156), lack of on-site testing capabilities 68% (106/156), lack of trained work force 50% (78/156), lack of awareness 41% (64/156), low prevalence of actionable PGx variants 38% (60/156) and lack of evidence 29% (46/156). In contrast, other clinicians reported that the lack of evidence 51% (79/156) and lack of professional awareness 43% (68/156) were either not a barrier or unlikely to be a barrier for the implementation (Figure 3a).
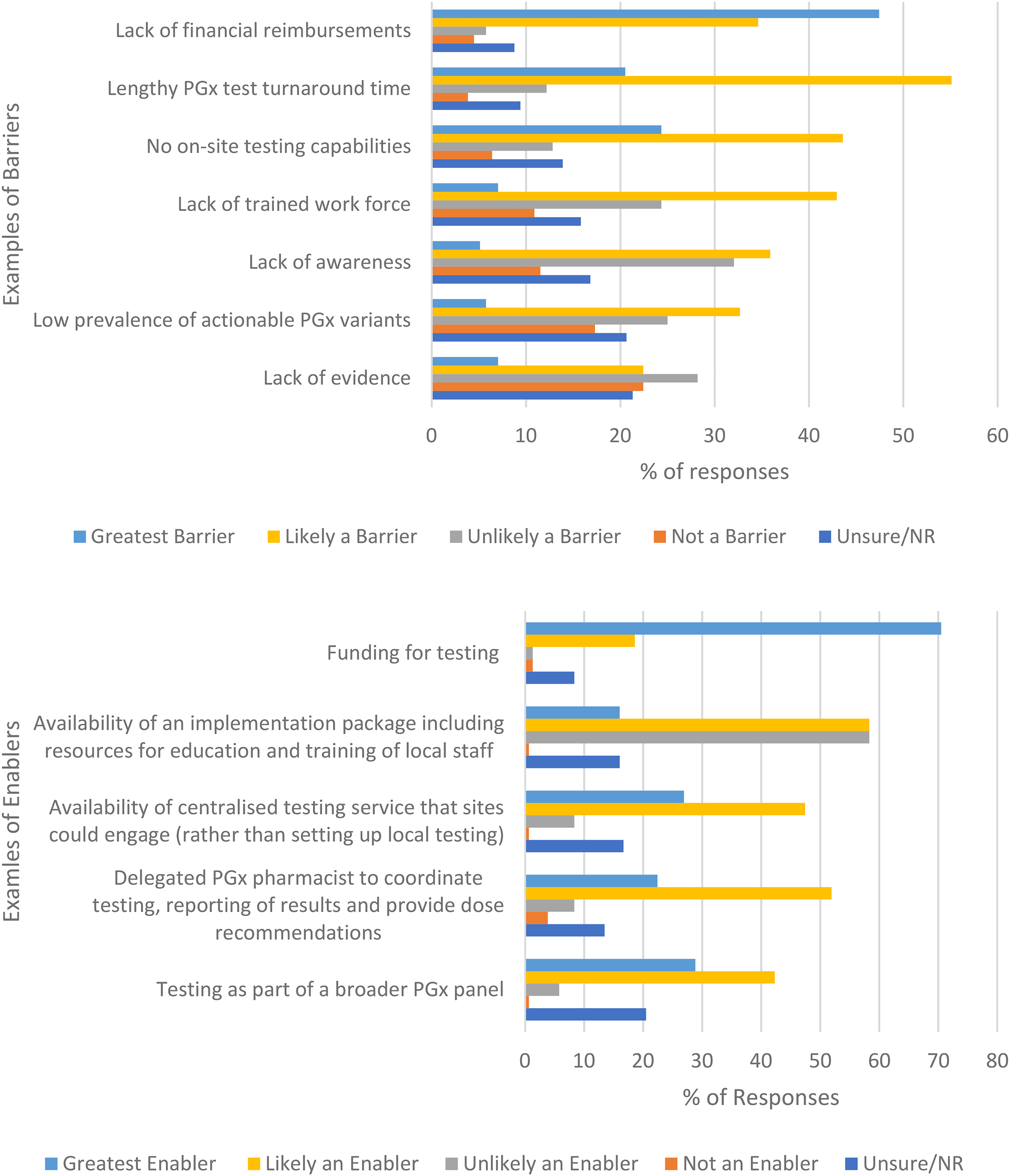
a. Clinician reported impact of potential barriers to implementation of Pharmacogenetic testing into routine clinical care. Abbreviations: NR: Not recorded; % percentage. b. Clinician reported impact of potential enablers to implementation of Pharmacogenetic testing into routine clinical care. Abbreviation: NR: Not recorded; % percentage.
Financial reimbursements were identified by 89% (139/156) of respondents as the greatest or likely enabler. Most clinicians identified the following enablers to implementation 1) a delegated PGx pharmacist to coordinate testing, reporting of results and provide dose recommendations 74% (116/156), 2) availability of an implementation package including resources for education and training of local staff 74% (116/156) and 3) the availability of centralised testing service that sites could engage (rather than setting up local testing) 74% (116/156) (Figure 3b). However, 58% (91/156) of the respondents have selected unlikely be an enabler to the availability of an implementation package including resources for education and training of local staff.
Other comments
When provided the opportunity for free-text response and comments related to any aspect of the survey or PGx screening, consistent themes were reported (Supplementary Material 1). Clinicians practicing in Australia reinforced survey response data for the importance of government funding or rebates to enable routine clinical implementation. There was also recognition of clinical need and disparity of access in the Australian context currently reliant on institution and/or patient self-funding programs. Comments included: ‘wish it was available at my cancer centre’ and ‘time to just do it’. Other comments detailed additional barriers not previously detailed in prior survey questions: 1) non-standardised reporting of results between providers causing confusion; 2) concern of creating delays to treatment delivery; 3) poor positive and negative predictive value of the test.
Discussion
This study represents one of few international real-world practice surveys, and the first in the Australian healthcare setting to assess the perspective of clinicians on pre-treatment DPYD and/or UGT1A1 gene testing. It highlights barriers and enablers to implementing a PGx program within routine clinical care. We provide contemporary data to show that, despite the known clinical benefits of pre-treatment DPYD and/or UGT1A1 in reducing serious adverse events, it has not been widely adopted.
This survey showed that only 21% of clinicians testing for DPYD as part of routine care and 12% as part of a clinical trial. These results were similar for the total responder group and when limited to Australian practitioners. These results are consistent with US data from a 2022 study where only 20% of oncologists reported having ever ordered testing before FP treatment and only 3% routinely order in their practices. 11
Our data has demonstrated that clinicians (77%) valued DPYD screening more than UGT1A1 (53%) may reflect hesitancies due to the lack of irinotecan specific drug target exposure levels or uniform recommendations between oncology, pharmacology societies and regulatory agencies in different countries. 6 Nevertheless, 84% of clinicians indicated that they would reduce irinotecan dose in patients with UGT1A1 poor metabolisers. Evidence from four meta-analyses show that patients who are homozygous to UGT1A1*28 have two-to four-fold increased risk of grade ≥ 3 neutropenia,12–15 and two-to six-fold increase in grade ≥ 3 diarrhoea,14–17 in comparison to heterozygous (*1/*28) or wild type (*1/*1). In a prospective study conducted by Hulshof 2022, 18 proved that UGT1A1 genotyping is cost effective and significantly reduces toxicity such as febrile neutropenia in UGT1A1 poor metabolisers in comparison to historical cohorts (6.5% versus 24%). 18 The systematic exposure of the active metabolite of irinotecan (SN-38) in UGT1A1 poor metabolisers received 30% dose reduction was slightly higher in comparison to patient cohort receiving full irinotecan dose. 18
DPWG guidelines recommend a 30% upfront dose reduction at cycle 1 for patients who are homozygous to UGT1A1*28, followed by dose increase from cycle 2 based on neutrophil count. 3 It is unlikely that upfront dose reduction to negatively impact the overall survival, as the recommended genotyping guided dosing leads to equal systemic exposure to SN-38 as in the wild type treated with standard irinotecan dose. 19
Most of clinicians were more comfortable to reduce (94%) or avoid (90%) FP doses in IM or PM patients receiving palliative intent treatment, given that symptom management and quality of life are the focus in patients’ goals of care. However, clinicians were hesitant to reduce (79%) or avoid FP (68%) in IM or PM patients receiving curative treatment. This hesitancy may reflect the concern that a dose reduction would negatively impact treatment response and outcomes especially where treatment intent is curative. Similar responses were reported in a US qualitative study 64% (n = 25) of oncology clinicians conducted by Lau-Min et al. 2022. 20 This could be due to major organisations such as National Comprehensive Cancer Network (NCCN) colon cancer guidelines panel does not recommend/support pre-emptive DPYD genotyping because of the risk of under dosing in DPYD variant allele carriers affecting their treatment response. 11 However, CPIC and DPWG guidelines recommend using an initial lower dose at cycle 1 and cycle 2, with up titration of dose in subsequent cycles based on tolerability to reduce the risk for under dosing.1,2 In line with these recommendations, a pharmacokinetic analysis conducted by Henricks et al., showed that 5-FU exposure in the DPYD variant allele carriers who were dose reduced was equal to the 5-FU exposure observed in the wild type: demonstrating that pre-treatment DPYD genotyping will not result in under dosing, and thus impacting overall response. 7
Glewis et al. addressed concerns that is evident in the survey data and published literature, by undertaking a systematic review and meta-analysis of studies that showed that PGx improves patient outcomes in terms of high grade toxicity, without impacting on treatment response. 4 However, we acknowledge limitations of these retrospective data in the systematic review and meta-analysis and the need for further prospective studies to definitively answer this question and to inform optimal clinical decision making. 4
In our study, 41% of clinicians indicated that lack of awareness and 39% thought that the evidence was lacking despite major organisations such as EMA, UK, FDA and Health Canada have assessed the available evidence as being sufficient to warrant their testing/dosing recommendations. These organisations warn against the use of FP in patients with absent DPD activity, 4 implying that there is no FP dose has been proven safe enough in patients with absent DPD activity. 21 A randomised control trial for DPYD gene testing (Boisdron-Celle et al.) with two cohorts (pre-treatment DPD deficiency screening versus control arm with no screening), was prematurely closed for ethical reasons. As a result of a patient's death in the control arm occurred due to DPYD PM status that discovered retrospectively. 22 This was evident in a meta-analysis conducted by Sharma et al. 2021, showing patients carrying pathogenic DPYD gene variants receiving a full dose of FP had more than 25 times increased risk of FP-treatment-related mortality. 23
Low sensitivity of PGx tests could be another reason for the hesitancy around dose reduction in curative treatment settings. The four common CPIC DPYD variants are more common among Caucasians than other ethnic populations. The test sensitivity and specificity shows greater or equal to 99% using Polymerase Chain Reaction (PCR) based for DPYD genotyping. 9 EMA in April 2020 recommended phenotype testing as an alternative method for DPYD genotyping, by measuring the uracil levels in the plasma prior to treatment to identify patients with DPD deficiency. 21 It is now used in European countries;24,25 however, concerns around pre-analytical issues and non-standardised reporting of results limit widespread implementation in clinical practice. 26 Therefore, a clinical PGx program that monitors patients for early actuate toxicities after first exposure will help minimise the impact of a low negative predictive value associated with DPYD-genotyping, until more evidence on other gene variants and metabolic pathways affecting FP-toxicity for a multiethnic global population becomes available.
In this survey, financial reimbursement for the PGx test was the most common factor preventing PGx testing with majority of clinicians agreeing that if made available, it will be the greatest enabler or likely an enabler to implementation. In comparison to the survey conducted in US oncologists, it wasn’t their first common and important factor as the clinicians were aware that DPYD testing is cost-saving, in comparison to the cost for the hospital admission for severe toxicity. 11 Currently DPYD testing is reimbursed by US Medicare and by some private insurance providers. 11 However, increasing insurance coverage would further increase the acceptance of DPYD testing across the US. 11 In Australia, there is no funding mechanism for DPYD gene testing, and the cost of hospital admission and chemotherapy treatment is covered by the public health system, making it less appealing for clinicians to refer patients for DPYD gene testing.
Perceptions of lengthy PGx test turnaround time was the second most common factor preventing clinicians to refer patients for pre-treatment PGx. Clinicians reported acceptable waiting times of five to seven business days, which is likely achievable 27 and therefore not a true barrier with contemporary testing capabilities. It was also one of the barriers identified by a Netherland study (Martens et al. 2020) in a semi-structured interview with pharmacists, oncologists and patients. 28 Similar results were reported by USA clinicians in gastrointestinal cancer settings, with the concern that many patients are very ill, requiring urgent treatment and the PGx test turnaround time should not take longer than one week, to become feasible. 20 These data show the importance of ensuring that pathology centres can feasibly deliver fast turn-around timing for gene test results in clinical practice. Centralised testing models may expedite testing capabilities for smaller and more remote centres, and should be evaluated in prospective studies.
Lack of on-site testing capabilities, lack of trained work force and low prevalence of actionable PGx variants were other common barriers highlighted in this survey. Clinicians indicated in this survey that by having 1) a dedicated program coordinator (we specified example of PGx pharmacist) to coordinate testing, reporting of results and provide dose recommendations, 2) availability of an implementation package including resources for education and 3) training of local staff and availability of centralised testing service that sites could engage (rather than setting up local testing) would be the greatest enabler or likely an enabler for the implementation of PGx services. Oncology trained pharmacists who meet competency requirements for PGx are well positioned to lead such programs for cancer care, given their background expertise in medicine disposition and clearance, medicine-medicine interactions and involvement in clinical verification of oncologist prescribed anti-cancer treatment plans and assessment of dose-adjustments. Our group is currently undertaking a multi-centre prospective study coordinated by a clinical PGx pharmacist to assess and evaluate the feasibility of implementing pre-treatment PGx screening for DPYD and UGT1A1, including a centralised model and follow-up of patients at metropolitan and regional centres (PACIFIC-PGx, ACTRN12621000251820). This will provide some of the feasibility data noted in the survey to be a current barrier to use.
A limitation identified in this study is the true denominator of individuals that the survey was emailed to, is unknown but we have been highly conservative presenting response rates as total distribution email lists given a significant overlap between the organisations.
PGx testing prior to FP and/or irinotecan is not routinely practiced despite evidence demonstrating that it would change clinical decision making in both curative and palliative treatment settings. Clinician hesitancy to reduce dosing for curative treatments was evident. Supportive data, education and implementation programs may overcome hesitancy and barriers to routine clinical utilisation.
Supplemental Material
sj-docx-1-opp-10.1177_10781552231167554 - Supplemental material for Pharmacogenetics testing (DPYD and UGT1A1) for fluoropyrimidine and irinotecan in routine clinical care: Perspectives of medical oncologists and oncology pharmacists
Supplemental material, sj-docx-1-opp-10.1177_10781552231167554 for Pharmacogenetics testing (DPYD and UGT1A1) for fluoropyrimidine and irinotecan in routine clinical care: Perspectives of medical oncologists and oncology pharmacists by Sarah Glewis, Senthil Lingaratnam, Mei Krishnasamy, Jennifer H Martin, Jeanne Tie, Marliese Alexander and Michael Michael in Journal of Oncology Pharmacy Practice
Footnotes
Author's Contributions
SG performed writing of the original draft and conceptualization. MK, SL, JHM, JT, MA and MM performed writing, review and editing; conceptualisation. Authors had full access to all of the data (including statistical reports and tables) in this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Ethics approval and consent to participate
The study was approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee ((HREC/80543/PMCC-2021). A decision to click on the survey link by the participants and to complete/submit the survey was taken as informed consent. The study was performed in accordance with the Declaration of Helsinki.
Consent for publication
N/A.
Data availability
N/A.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.