
Abstract
Background
Factor XI (FXI) deficiency is a rare bleeding disorder, common among Ashkenazi Jews. Affected individuals may present severe bleeding after trauma or surgery, which requires careful management. To date FXI deficiency has not been genetically or clinically described in the Jordanian population.
Material and methods
Seventeen patients (median age 29) from 16 Jordanian Arab families, referred to the hemostasis and thrombosis laboratory at the University of Jordan, due to bleeding history or prolonged activated partial thromboplastin time. FXI deficiency was confirmed by factor assay (cutoff: ≤ 40% activity). Bleeding score was recorded, and Next Generation Sequencing of the F11 gene was utilized to detect causative mutations.
Results
Of the 17 patients, 14 had severe FXI deficiency. The median bleeding score was 6. Five patients carried the known p.Glu135Ter (type II), while ten patients had different point mutations. Three patients with severe FXI deficiency were found to carry previously undescribed variants, including one novel splice site mutation (c.1136-1G > C). No pathogenic variants were identified in two patients.
Discussion
This is the first study of FXI deficiency in Jordanian Arabs, which revealed fourteen patients with severe FXI deficiency. Ten mutations were identified, including one novel splice-site mutation. The relevance of type II mutation in our cohort suggested a founder effect similar to Jewish ancestry. Bleeding severity was incongruent with FXI activity. Despite the small cohort and lack of functional assays, this study expands the mutational spectrum and provides new insights into FXI deficiency in Arab populations.
Keywords
Introduction
Coagulation Factor XI (FXI) is a 160-kDa glycoprotein originally made in the liver cells and secreted into the blood as a zymogen of Factor XIa. It acts as a serine protease within the coagulation cascade, playing an important role in the activation of Factor IX, which is a key step in thrombin generation at sites of injury. 1 Factor XI deficiency is a rare congenital bleeding disorder that was first identified and comprehensively described in Ashkenazi Jews, with subsequent reports in other ethnic groups.2–4 The most predominant mutation in individuals of Jewish ancestry is p.Glu135Ter, which is defined as Type II based on the mechanistic classification system for FXI deficiency, and to date, according to the Factor XI gene (F11) database, 272 DNA variants have been registered.5,6
Patients with Factor XI deficiency are at increased risk of bleeding, especially during or after surgeries, where heavy bleeding can be life-threatening. In women, pregnancy requires cautious management, although miscarriage is not ordinarily seen with this condition. 7 The prevalence and genetic basis of Factor XI deficiency vary considerably across populations. For example, the Cys38Arg mutation has been reported in eight families of French Basques, with variations in zygosity patterns. 8 Similarly, different mutations have been identified in non-Jewish populations in the UK,9–11 as well as in East Asian populations, including Japan, China, and Korea.12–14
The severity of Factor XI deficiency is classified based on plasma FXI activity levels as severe deficiency (< 20%), while mild deficiency (20% - 40%), as reported previously. 15
Some studies have reported the prevalence of Factor XI deficiency without providing genetic analysis in Arab populations such as Tunisia, 16 Oman, 17 and Saudi Arabia. 18 In Jordan, Factor XI deficiency was first clinically observed in 1984, as mentioned, 19 but since then, no detailed family-based genetic studies have been conducted in the Jordanian population.
Our study aims to identify causative mutations in the F11 gene that cause Factor XI deficiency in affected Jordanian patients and their families, to correlate these mutations with bleeding scores, and to identify the relationship between the probable effects of specific mutations on plasma FXI levels and bleeding severity. Our results revealed ten different mutations, the most common is the stop-gained mutation
Materials and Methods
Subjects
This study included 16 unrelated families, along with their affected members, except for three patients who were tested without their families. All patients were from the Jordanian population and non-Jewish origin. A total of 17 patients (6 males and 11 females), while one family had two siblings with Factor XI (FXI) deficiency: a male proband and his sister. All patients were referred from the Thrombosis and Hemostasis Laboratory at the University of Jordan, following clinical suspicion of FXI deficiency, with most cases diagnosed after noticing prolonged activated partial thromboplastin time (aPTT) as a preoperative test.
Laboratory assessments included prothrombin time (PT), activated partial thromboplastin time (aPTT), thrombin time (TT), and coagulation factor assays (FVIII, FIX, and FXI) performed according to previously published methods.19,20 Notably, none of the patients were on anticoagulant therapy at the time of testing, except one patient who took anticoagulant therapy after heart valve replacement. A mixing study was performed to rule out the presence of inhibitors that may affect the coagulation cascade. Bleeding severity was evaluated using the International Society on Thrombosis and Haemostasis Bleeding Assessment Tool (ISTH-BAT). 21 Abnormal bleeding scores were defined as ≥6 for females and ≥4 for males, based on the ISTH-BAT questionnaire.
Laboratory Clot-Based Tests and Factor Assays
Citrated plasma samples were tested for Prothrombin Time (PT), Activated Partial Thromboplastin Time (aPTT), Thrombin Time (TT), mixing study for aPTT, and Coagulation Factor Assays (FVIII, FIX, FXI, and FXII), were done manually as previously described. 19 Severe FXI level activity was considered less than 20%.
DNA Extraction and PCR
Whole blood was collected from 4 mL EDTA-anticoagulant tubes, and DNA was extracted using a commercial kit (Qiagen MinElute, Germany) following the manufacturer's protocol. Concentration of DNA and purity were measured using a spectrophotometer (NanoDrop 2000, USA), with an acceptable concentration of 50 ng/μL and purity around 1.89 at 260/280 nm absorbance.
Initial screening for the known Type II founder mutation (c.403G > T, p.Glu135Ter) was performed using Sanger sequencing. Patients who tested negative for this mutation underwent additional analysis using whole-exome sequencing to detect other F11 variants. Primers were designed with Primer-BLAST (NCBI) and Primer3 software (ELIXIR) (
Primers Sequences Used in This Study.

DNA Sequencing
Sanger sequencing was performed using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA). Bidirectional sequencing products were purified using the NucleoSEQ kit (MACHEREY-NAGEL, USA), and capillary electrophoresis was performed using ABI 3500 Genetic Analyzer (Applied Biosystems, USA). Sequencing files were aligned against the reference transcript (ENST00000403665.7) using Chromas Pro (Technelysium, Australia). Detected mutations were analyzed using Ensembl Genome Browser (EMBL-EBI, Cambridge, UK), available at https://www.ensembl.org/ (accessed on 1/1/2025), and ClinVar archive maintained at the National Institutes of Health (NIH) (Bethesda,USA), available at https://www.ncbi.nlm.nih.gov/clinvar/. (accessed on 1/1/2025). The variant annotation was performed using ACMG classification. 22
Next-Generation Sequencing
Samples negative for the Type II in exon 5 during initial screening underwent further analysis using next-generation sequencing (NGS). Libraries were prepared using the DNA Prep with Enrichment kit (Illumina, USA) and sequenced on NextSeq 550 system (Illumina, USA). Data analysis was performed using Variant Interpreter https://variantinterpreter.informatics.illumina.com/, (accessed on 1/12/2024). BAM and BAI files were visualized using Integrative Genomics Viewer https://igv.org/app/ (accessed on 1/12/2024) to confirm the reading alignment and depth before segregation analysis.
Results
Bleeding Severity Assessment
Bleeding severity was assessed using the ISTH-SSC Bleeding Assessment Tool.
21
Several patients experienced bleeding episodes for the first time during surgical procedures. The median bleeding score and the dominant site of bleeding as shown in (
Summary of Patients’ Bleeding Score with Dominant Site of Bleeding.
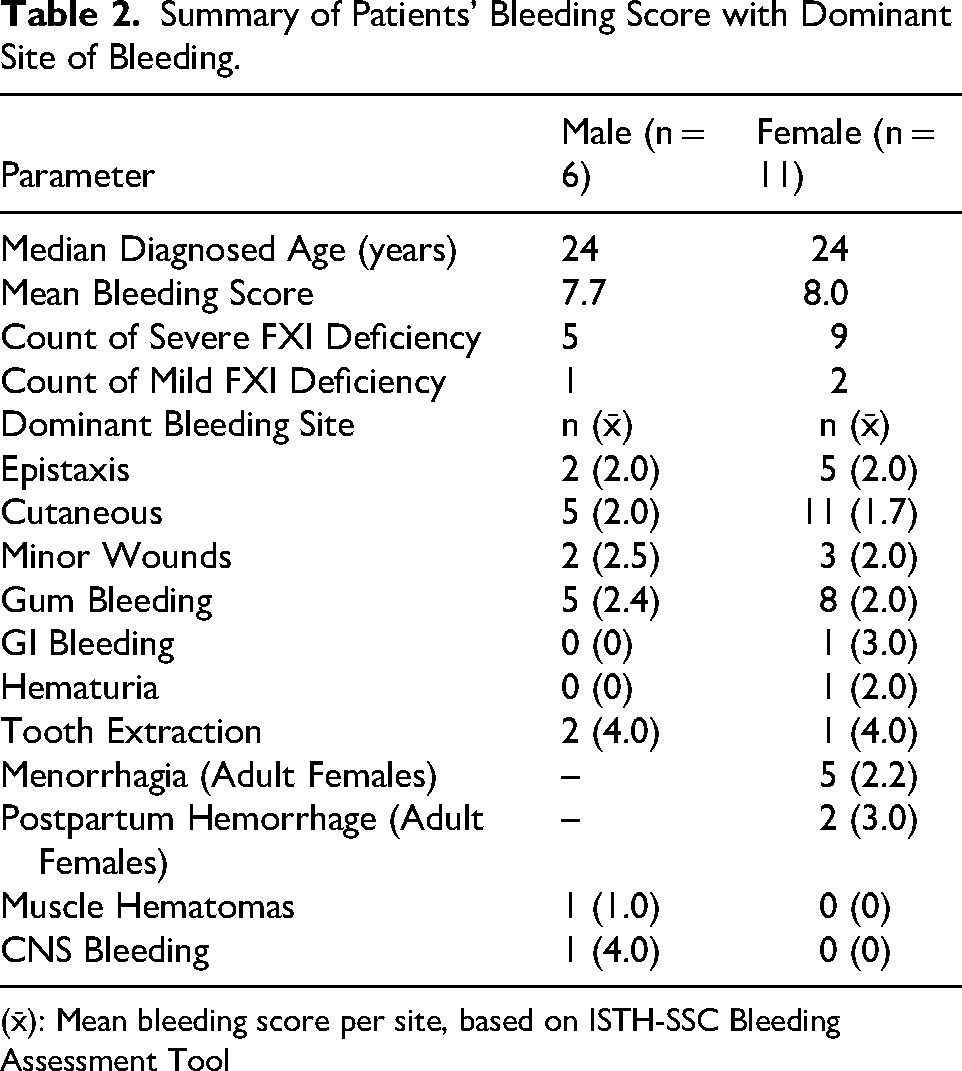
(x̄): Mean bleeding score per site, based on ISTH-SSC Bleeding Assessment Tool
Clot-Based Activity Assay
Results for PT were within the normal range for all patients except for one case, whose recorded slightly high PT (30 s) because of anti-coagulant therapy post-heart valve replacement. Results for TT were normal across all samples. Results for aPTT were prolonged (> 40 s) for all patients, consistent with FXI deficiency. Some patients exceeded two minutes and were recorded (> 120 s). Mixing studies with normal pooled citrated plasma were done and showed correction of aPTT for all patients, ruling out internal inhibitors. The three patients of particular interest, including the one carrying the novel mutation, exhibited prolonged aPTT values (>120 s) (Table 3).
Genotype–Phenotype Correlations in FXI Deficiency Patients in This Study.

†Patients requiring further investigation beyond FXI deficiency.
‡Protein changes are shown in a shortened format. Full HGVS nomenclature is provided in the manuscript text.
§FFP, req, indicates patient regularly receives fresh frozen plasma.
Abbreviations: ΔaPTT, seconds above control value; BS, bleeding score; AS, asymptomatic; BT, blood transfusion; CCY, cholecystectomy; CNS, central nervous system bleeding; Co, compound heterozygous; Cut, cutaneous bleeding; Ep, epistaxis; GI, gastrointestinal bleeding; G, gum bleeding; H, hematuria; HEM, hemorrhoidectomy; HS, hemorrhagic stroke; M, menorrhagia; MC, miscarriage; Mi, minor; PPH, postpartum hemorrhage; PS, phenotype severity; S, severe; TE, tooth extraction; Tons, tonsillectomy; CS, cesarean section; CE, cervical ectropion; ENT, ear, nose, and throat surgery; BTS, brain tumor surgery; VR, valve replacement; Hyst, hysterectomy; Sept, septoplasty.
Coagulation Factors Activity Levels
Our findings revealed that 14 out of 17 patients (82.3%) exhibited severe Factor XI (FXI) deficiency, defined as plasma FXI activity below 20%. Ten out of these 14 patients (71.4%) had very severe deficiency of less than 10%. In contrast, 3 out of 17 patients (17.6%) appeared to have mild deficiency, with FXI activity ranging between 20% and 40%. Coagulation factors FVIII and FIX levels within the normal range; however, patients (A/1and O/4) showed low FVIII activity, patient (K/1) showed low FIX activity, and patients (B/1, H/1, and J/1) showed reduced levels of both FVIII and FIX activity; further studies should be done to explore the reasons beyond FXI deficiency
Genetic Findings
Our results identified that 5 out of 17 patients (29.4%) were homozygous for the c.403G > T (p.Glu135Ter) mutation (Type II) in exon 5. Ten patients out of 17 harbored various mutations in the F11 gene, predominantly clustered in the serine protease domain. The remaining two patients had no identified pathogenic mutations
F11 gene sequencing
Sequencing of the F11 gene identified ten distinct point mutations: one stop-gained mutation, one novel splice site mutation, and eight other missense mutations. In silico predictions were checked to understand their effect on FXI level. Two variants (c.1016G > T,c.731A > G) were classified as benign according to ACMG guidelines, despite being registered in the EAHAD database in patients with FXI deficiency. However, in our study, these variants were detected only in heterozygous form. All patients, except three, were tested along with their family members. All familial segregation analysis is shown in (

Pedigrees of 11 Families Showing FXI Deficiency. (A) Family. A Shows Mutation Glu135Ter (Type II) in Homozygous State for one Female Patient and Segregated Within her Family Members in Heterozygous State. (B) Family C Shows Mutation Glu135Ter (Type II) in Homozygous State for one Male Patient with Segregation in Heterozygous State for his Family. (C) Family D Shows Glu135Ter (Type II) in one Homozygous State for one Female Patient and Segregated Within her Family Members in Heterozygous State. (D) Family F Shows Mutation Arg326His for Affected Proband in Homozygous State and her Father in Heterozygous State. (E) Family G Shows Mutation Ile421Thr in Homozygous State for the Patient and Segregated in His Family. (F) Family H Shows Mutation Leu532Pro in Homozygous State for the Patient and Appeared in Heterozygous State for his Affected Sister and Other Family Members. (G) Family J Shows Mutation. (H) Family K Shows Mutation Cys339Phe. (I) Family L Shows Mutation Tyr608His. (J) Family M Shows Mutation Gly354Arg Compound Heterozygous with Tyr608His in one Female Patient and no one of her Family Members Were Compound. (K) Family N Shows Glu620Gln as Heterozygous for the Patient and his Mother Only.
c.403G > T (p.Glu135Ter) Mutation
This mutation was found in five patients from five unrelated families (A, B, C, D, and E) in a homozygous state. These patients suffered from severe bleeding phenotypes, which required high numbers of fresh frozen plasma (FFP) transfusions during their bleeding episodes, as shown in
Other Genetic Findings
Discussion
The outcomes of this study regarding Factor XI (FXI) deficiency provide precious insights into the genetic and clinical characteristics of this rare bleeding disorder in Jordan and perhaps in the Arab population.
In this study, 82.3% of patients exhibited severe FXI deficiency (FXI activity <20%), with 71.4% of these cases classified as very severe (FXI activity <10%). This distribution is consistent with reports from other populations, such as Ashkenazi Jews, where severe FXI deficiency is mostly attributed to founder mutations like c.403G > T (p.Glu135Ter).2,3 However, the proportion of severe cases in the Jordanian families is higher than in some other populations, considering that the Jordanian population is non-Jewish origin. The c.403G > T (p.Glu135Ter) mutation was identified in 29.4% of our patients. The high prevalence of this mutation in the Jordanian group suggests a possible founder effect, while other populations have other founder effect mutations such as French Basques. 8 In some populations, such as the UK, milder forms are more common.4,10 This discrepancy may be considered genetic heterogeneity or founder effects related to the Jordanian population. Our findings are also consistent with studies from China, Korea, and Italy, which have reported other population-specific mutations.12,13,24
Additionally, the identification of ten mutations, including novel mutation c.1136-1G > C (splice site mutation) that is not reported before for FXI patients that seems to have RNA splicing, suggested that it may disrupt the consensus splice site, which can lead to exon skipping and has a pathogenic effect.25,26,31
The bleeding severity observed in our cohort, as assessed by the ISTH-SSC Bleeding Assessment Tool is in line with global reports. Menorrhagia (50% of female patients) and postpartum hemorrhage (20%) were common, consistent with findings from the UK and other regions.7,15 However, the high incidence of severe bleeding episodes that required fresh frozen plasma (FFP) transfusions in patients with the c.403G > T mutation highlights the clinical severity of this genotype, consistent with reports from Ashkenazi Jewish populations.2,3
Our work has identified a wide range of F11 gene mutations, including a novel one, engaged to the global recognition of FXI deficiency. Through familial segregation analysis, we strengthened the interpretation of these genetic findings and clarified the significance of variants with previously uncertain effects.
The combination of bleeding scores, coagulation factor assays, and DNA sequencing data provided a comprehensive overview of the disorder in our cohort and may help expand understanding in similar populations within the region.
The high proportion of severe FXI deficiency in this study, particularly among patients with the c.403G > T mutation, suggests a unique genetic burden in the Jordanian population, while the novel and all other genetic findings can give an added value to the database across worldwide.
The primary limitation of our study is the small sample size comprising 17 patients, which minimizes the generalizability of our findings. Larger cohorts are needed to confirm the prevalence and clinical significance of the identified mutations.
This study lacks the exploring RNA splicing that would provide deeper knowledge into the molecular mechanisms underlying FXI deficiency.
Another limitation is the absence of in vivo functional analysis to assess the impact of newly reported variants on the FXI protein.
The importance of this work lies in its provision of clear insight into the clinical and genetic aspects of FXI deficiency within the Arab population.
Conclusions
This study provides valuable data into the genetic and clinical characteristics of FXI deficiency in Jordanian families. The high prevalence of severe deficiency and the identification of novel mutation underscore the importance of population-specific studies to understand the genetic diversity of this disorder. We recognize the need to expand the study to engage more patients from other countries in the region.
Footnotes
Acknowledgments
The authors would like to acknowledge the support of the Deanship of Scientific Research at the University of Jordan.
Ethical Considerations
The work was approved by the institutional review board of Cell Therapy Center, Amman, Jordan (CTC) [IRB-CTC/1-2021/03]. All research activities were conducted in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Consent to Participate
Written informed consent was obtained from all participants or their legal guardians prior to sample collection and genetic analysis.
Author Contributions
Conceptualization: FF, AA
Methodology: LM, SA, AA
Data curation: FN, FF, AS, SH, ON, LA, TA
Writing – original draft preparation: SA, AA, FF
Writing – review & editing: SA, AA
Project administration: AA, SA, FF, AS, SH
Funding acquisition: AA, LM
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Deanship of Scientific Research of the University of Jordan (grant No. 2022-2021/8).
Declaration of Conflicting Interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare no financial or personal conflicts of interest.
Data Availability
The datasets analyzed during the current study are not publicly available due to ethical restrictions, but are available from the corresponding author on reasonable request.