
Abstract
Hemophilia B (HB) is an inherited bleeding disorder caused by defects in the FⅨ gene, leading to severe coagulation dysfunction. This study designed eight pairs of primers covering eight exons of the FⅨ gene and used PCR and DNA sequencing to detect FⅨ gene mutations in 31 HB patients. Sequencing results were compared with normal sequences using Chromas software on Blast to identify mutation sites. Findings revealed the CpG dinucleotide region as a mutation hotspot and the 192nd nucleotide (FⅨ192) as a dinucleotide polymorphism site in the Chinese population. Pathogenic mutations included point mutations, deletions, insertions, and mutations affecting amino acids or splicing sites. For cases with only polymorphic sites, further exon sequencing is needed. This study adds new mutation data to the global HB database, supports research on racial differences in FⅨ gene mutations, and contributes to domestic HB statistics. The results aid in understanding the FⅨ gene's role in coagulation, elucidating HB pathogenesis, and providing a basis for future gene therapy.
Introduction
Haemophilia is a group of inherited haemorrhagic disorders caused by defects in certain clotting factors in the blood, leading to severe coagulation difficulties. It can affect both men and women, but most patients are men. 1 The main types of haemophilia are haemophilia A (hemophilia type A), haemophilia B (hemophilia type B), factor XI deficiency (also known as haemophilia C), and other factor deficiencies. Haemophilia is the most common of the congenital haemorrhagic disorders, and bleeding is the main clinical manifestation. The most common clinical haemophilias are haemophilia A (HA) and haemophilia B (HB), which are caused by defects in the genes for coagulation factor VIII (FⅧ) and coagulation factor IX (FⅨ), respectively. These are both X-linked recessive inherited diseases. In male haemophilia patients, HA accounts for 80% to 85%, while HB accounts for only 15% to 20%. HB has a prevalence of approximately 1/30,000 in men, which is much lower than that of HA, and female patients are rare. 2 The current main treatment for haemophilia is to prevent or control haemorrhagic symptoms by supplementing the body with coagulation factors and other plasma replacements. As hemophilia is a genetic disease, it is necessary to treat the corresponding mutated gene to reduce the mortality rate. Therefore, it is necessary to further study and analyze the gene mutation types of this disease to establish an effective genetic diagnosis system and provide important evidence for gene therapy.
Hemophilia B, also known as Christmas Disease, is caused by the defect of FⅨ gene, leading to the decrease or dysfunction of FⅨ in plasma and the reduction or loss of coagulation activity of coagulation factors, thus forming severe coagulation dysfunction. 3 In 1985, Yoshitake et al completed the full sequence analysis of the FⅨ gene. 4 It can be known that the length of the FⅨ gene is 33.5 kb, which is composed of 8 exons, 7 introns and flanking sequences, and its 8 exons encode the 6 domains of FⅨ protein. In 1990, George Brownlee first established the HB gene mutation database. 5 The latest FⅨ Mutation Database has collected a total of 5358 patients, covering 1692 unique mutations. 6 With the deepening of HB research and the increasing number of HB patients worldwide, the mutation database is constantly being updated, and new mutations are being discovered and recognized. According to the distribution of mutations in protein regions and control regions within the database, the mutations of FⅨ gene have a high degree of heterogeneity, which can occur at any position of FⅨ gene exons, introns, promoters and flanking sequences. The known types of FⅨ gene mutations include missense mutations, nonsense mutations, small insertions and deletions, among which missense mutations account for more than 60%. 7
At present, the gene diagnosis methods for HB and carriers can be roughly divided into two categories: indirect method and direct method. 8 For families with clear family history and probands, multiple STR polymorphism loci can be combined to use indirect gene diagnosis methods such as restriction fragment length polymorphism (RFLPs), short tandem repeats (STRs), and single nucleotide polymorphism markers (SNPs) to diagnose HB. Direct diagnosis methods such as denaturing gradient gel electrophoresis (DGGE) and single-strand conformational polymorphism (SSCP) can be used by amplifying the target gene with PCR and then performing heteroduplex analysis. For HB cases without family history or probands and for carriers, even if the linkage analysis method using polymorphic loci is used, there may still be cases that cannot be diagnosed. When abnormalities are found by direct detection methods such as SSCP and DGGE, further gene sequence analysis is still needed to determine the specific mutations, and these methods have problems such as complexity, time-consuming, and low sensitivity. 9 In recent years, with the continuous maturity of PCR technology and other related factors such as the small FⅨ gene, many research groups at home and abroad have used whole-genome sequencing methods to replace other methods for the genetic diagnosis of HB patients, and have achieved good results. 10 Based on literature and other research, this study designed and synthesized 8 pairs of primers covering the 8 exons and flanking sequences of the FⅨ gene. The polymerase chain reaction (PCR) and DNA sequencing technology were used to detect FⅨ gene mutations in 31 HB patients. The sequencing results were compared with normal sequences on Blast by Chromas software to find the specific location of gene mutations.
All the gene mutations of FⅨ in HB patients collected by the study were directly sequenced, and the specific location, type and amino acid changes of the mutations were analyzed. The detection and analysis of gene mutations in 31 patients are of great significance for clarifying the mechanism of FⅨ gene mutations, providing molecular evidence for the development of gene therapy, and making necessary supplements to the world's HB mutation database. It is an important part of HB research worldwide.
Materials and Methods
Subjects
Thirty-one patients with hemophilia B (HB) from the hematology and obstetrics departments of Southern Hospital, with no known relatives among them, were selected. These patients all showed varying degrees of bleeding, and all had factor IX activity (FⅨ:C) of less than 5%.
Conventional PCR Reagents
Oligonucleotide primers: synthesized by Shanghai Yingjun Biotech Co., Ltd and purified by PAGE.
Conventional Taq DNA polymerase: from Bio Basic Inc. (Canada).
dNTPs: from Biobasic Inc. (Canada).
Specimen Collection and DNA Extraction
2 ml of peripheral blood was collected from HB patients in the vein, using ethylenediamine tetraacetic acid (EDTA) as the anticoagulant. In the laboratory, genomic DNA was extracted from the peripheral blood using a commercial kit (Life Technologies, USA) and stored at −20°C.
Primer Design
Eight pairs of primers were designed based on the sequence of the FⅨ gene (Gene Bank K02402) and literature references. The primers covered all of the exons and flanking sequences of the FⅨ gene. The primers were synthesized by Shanghai Yingjun Biotech Co., Ltd (Table 1).
Primers Sequence.
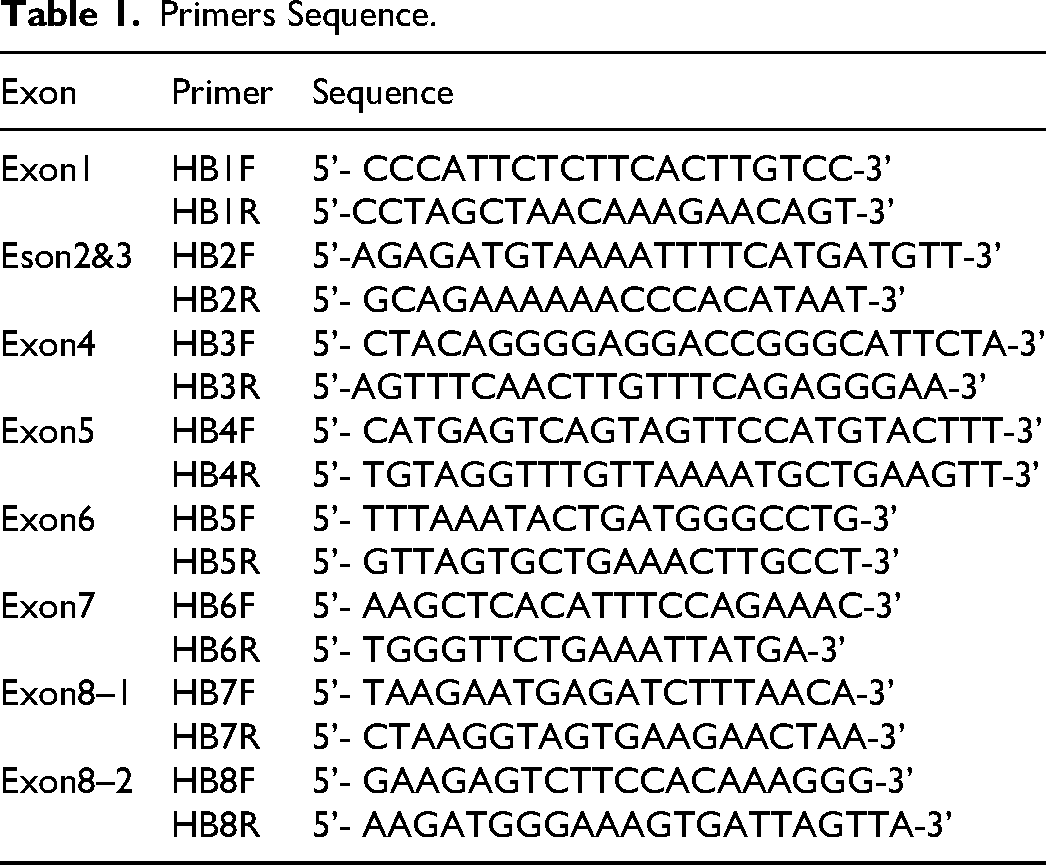
Primers Sequence.
The PCR reaction volume was 50 μl, containing 5 pmol of upstream and downstream primers each (final concentration of 0.1 μM), 5 nmol of each dNTP (final concentration of 0.1 mM), 5 μl of 10 × Taq PCR Buffer (100 mM Tris-HCl, 15 mM MgCl2, and 500 mM KCl, pH 8.3), 2 U Taq DNA polymerase (TAKARA), and 50–200 ng of genomic DNA. All PCR amplification reactions were performed on a thermal cycler (9700 model, PE Company). The PCR conditions for the eight pairs of primers were as follows: initial denaturation at 95°C for 5 min; 30 cycles of denaturation at 94°C for 30 s, annealing at 59°C for 30 s, extension at 72°C for 1 min; and a final extension at 72°C for 10 min.
Electrophoresis and Sequencing of PCR Products
Five microliters of the PCR amplification product were mixed with 1 μl of loading buffer, and electrophoresis was performed on a 1% agarose gel stained with ethidium bromide (EB). The electrophoresis was run at 80 V for 20 min to verify the specificity of the PCR amplification. The PCR products that met the sequencing conditions were sent to Shanghai Yingjun Biotech Co., Ltd, for purification and sequencing. The sequencing results were analyzed using Chromas and Blast bioinformatics software.
Sequencing Result Comparison Analysis
The sequencing results were compared with the normal gene sequence on Blaster using Chromas software to determine the specific location and base changes of the gene mutations.
Result
Thirty-one HB patients without family history were tested for mutations by PCR and DNA direct sequencing. All patients were found to have mutation sites. The detection results showed that the mutation sites of FⅨ gene were scattered, and the mutation types were diverse. The mutation types included point mutations, small fragment deletions and large fragment deletions, and point mutations accounted for the majority.
In this study, 31 patients were found to have 34 mutations, including one triple mutation (Case 15), six double mutations (Case 1, 4, 13, 17, 25, 26), and one case (Case 23) of whole gene deletion. Among the 34 mutations, 13 new mutations were found according to the latest mutation database, which were the first reported mutation types in the world.
The specific sequencing maps of the 13 newly discovered mutations are shown in the attached figure section (Fig. 1).

The specific sequencing maps of the 13 newly discovered mutations.
CpG is a mutation hotspot region. Among the 31 patients in this study, 7 patients had mutations at CpG dinucleotide sites. In addition, there were 4 patients whose mutations occurred at the 192nd nucleotide, and this site was a dinucleotide polymorphism site.
The base changes, specific positions (gene base sequence numbering using Yoshitake method), amino acid changes caused by gene mutations, and whether they were CpG mutation hotspots of 31 HB patients are listed in Table 2.
FⅨ Gene Mutation in 31 HB Patients.
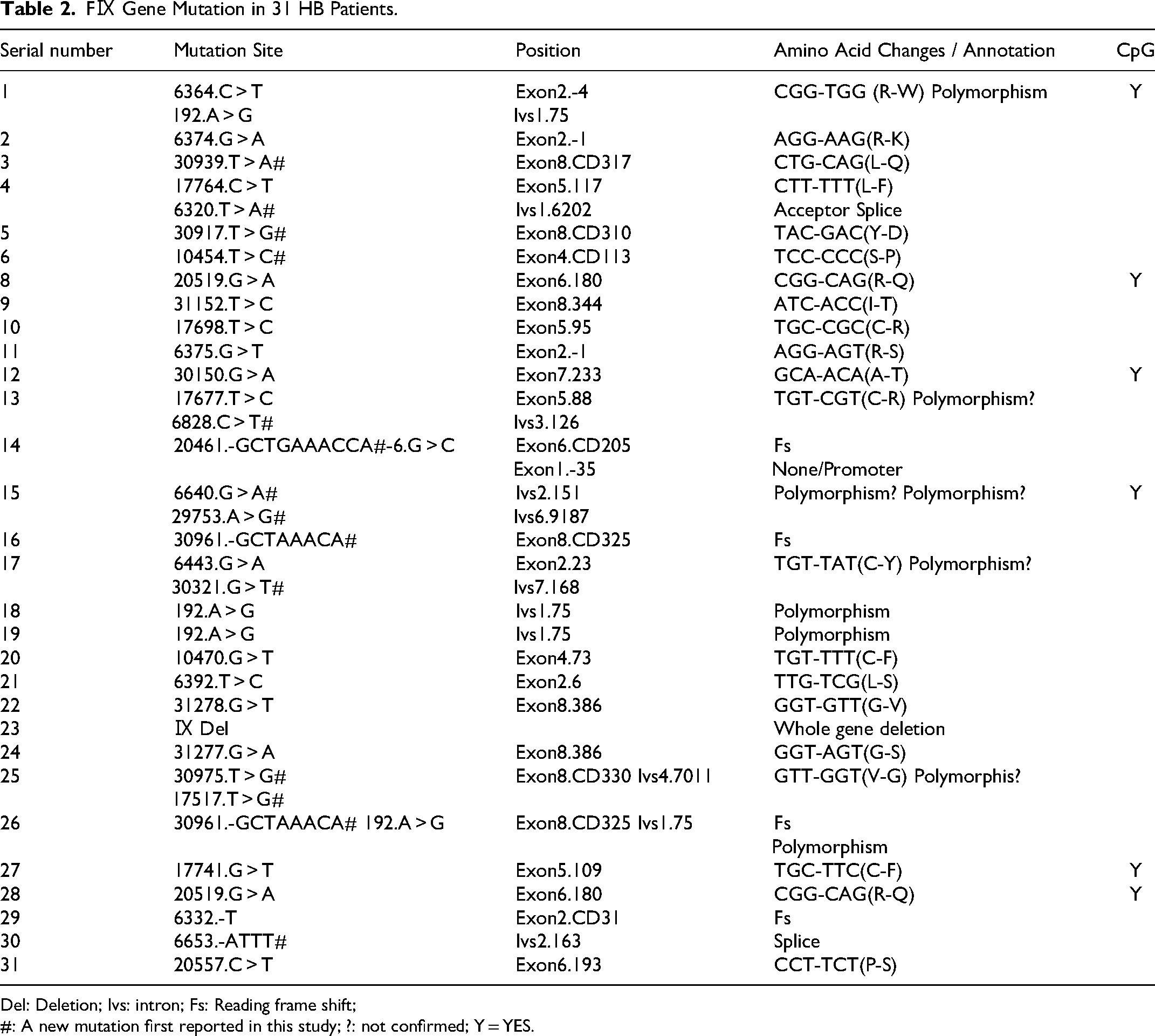
Del: Deletion; Ivs: intron; Fs: Reading frame shift;
#: A new mutation first reported in this study; ?: not confirmed; Y = YES.
The structure and function of FⅨ gene and its protein have been well studied so far. We know that the FⅨ gene is located at Xq27.1, and consists of 8 exons, 7 introns, and regulatory regions in the flanking sequences. The full-length mRNA is 2804 bp. The 8 exons encode 6 major domains of the protein, which are closely related to the normal function of FⅨ protein. 11 Gene mutations can cause conformational changes in the protein, reduced binding capacity with other factors, and reduced catalytic activity, and other serious consequences. Therefore, defects at any position of the FⅨ gene cause changes in the structure, function, and quantity of the protein, which can lead to the occurrence of HB. 12
There are various types of mutations in the HB gene, with point mutations, small-fragment deletions (less than 30 bp), and insertional mutations being more common, while large-fragment deletions (more than 30 bp) and insertions are rare. Among them, about 80% are single-base mutations. 12 Unlike HA, HB has a scattered occurrence rate of 30–50%. The latest FⅨ mutation Database has found a total of 1692 types of mutations. From the reported mutation distribution, mutations have occurred in all regions of the FⅨ gene, including the polyA signal. 6
With the progress of science and technology, people have more and more understanding of hemophilia, but there are still many conditions limiting the recognition of HB. First, the limitation of case number. HB is an X-linked recessive inherited disease. Although HB is also one of the most common types of hemophilia, the incidence of HB in the population is only 1/30000, and the ratio of HB to HA is about 1:5. The number of HB cases is very small. 13 Secondly, the study of HB gene mutation mechanism is also very limited. HB sporadic cases are common, and many cases have no family history or proband. There are also various types of gene mutations, and the mutation distribution covers all regions of the gene sequence. Whether many mutation loci cause the occurrence of HB or whether they are just polymorphic loci cannot be determined. 14 Again, so far, there is no a series of simple and rapid diagnostic methods. Many gene diagnostic methods for HB are either too cumbersome and time-consuming, or cannot locate the mutation sites specifically. The cases of familial hemophilia are even more difficult to make a final diagnosis. 10 Therefore, although the international HB gene mutation database has been established long ago, the database only contains thousands of mutation cases, and the mutation data it contains is also very limited.
Compared with international research, domestic research on HB is even more limited. There is still a huge gap between China's medical level and medical research and that of developed countries. Although China is a populous country, given the limited level of people's cognition of HB, the number of clinical cases of HB that are diagnosed and treated is very small. Therefore, there is a lack of detailed statistical data on the incidence of hemophilia in mainland China. 15 According to reports, the incidence of hemophilia in China is lower than that in Western countries, with HA at 1.95∼2.40/105 and HB at 0.32∼0.44/105. 16 In recent years, although some research institutions and researchers have begun to study HB, the available data on HB in the Chinese population is still very limited. The number of HB cases collected in this study has reached several tens of cases, which will provide very valuable research data for the study of HB, an important supplement to the data on HB gene mutations in the Chinese population, and also an important supplement to the research on HB worldwide.
In general, this study collected a total of 31 HB patients without family history. Excluding 6 cases of mutations (1 case of whole gene deletion mutation and 5 cases of short fragment deletion mutation), the rest were point mutations, accounting for 80.6% of the cases. It can be seen that point mutations are still the most common type of mutation. Point mutations in the coding region are prone to cause missense mutations, synonymous mutations, and nonsense mutations, which can lead to a series of abnormalities in protein transcription and translation, and then cause different levels of HB. 17 Among the deletion types, there is 1 case of whole gene deletion, which means there is no FⅨ production. The deletion of the FⅨ gene leads to the inability of the endogenous coagulation pathway to function normally, resulting in severe B hemophilia. When compared with the clinical bleeding degree of HB patients, it completely matches their genetic mutation types. 12
HB is clinically more common in mild cases, and the bleeding is lighter than HA. Among the 8 exons of the FⅨ gene, exons 4 and 8 encode regions that are very important functional areas because they encode regions that bind to Ca2+ with EGF and catalytic regions. Mutations in these two regions are prone to cause changes in protein structure or quantity, leading to different levels of B hemophilia. Therefore, the mutation rates of exons 4 and 8 are higher. 18 Although there were only 31 patients in this study, a total of 11 cases occurred in exons 4 and 8, accounting for 35.5%. In contrast, there were fewer cases of mutations that occurred in exons 1, 6, and 7 regions, accounting for only 19.4%. This may be because these regions encode proteins that play a less significant role. It can be seen that the distribution of exon region mutations in this study is roughly consistent with the overall HB mutation distribution.
After studying a large number of mutations in B hemophilia, the CpG dinucleotide region is considered to be a mutation hotspot. 19 Karl J Fryxell et al confirm that the CpG is indeed a mutation hotspot, with the main mutation being C → T/A. 20 In the study, 7 patients had mutations in the CpG hotspot, and the mutation types were C → T, G → A and G → T, accounting for 22.6% of the mutations. The mutations found in the CpG mutation hotspot not only verify again that this region is a mutation hotspot, but also provide new mutation type data in this region (G → A/T), which is an important complement to the mutation types in HB mutation hotspot regions.
The occurrence of gene mutations is different among human races, especially the polymorphism of genes. 21 Many gene diagnosis methods are based on the polymorphism of genes. Limited by the differences of human races in various regions and the immature experimental conditions, the research on the polymorphic loci of FⅨ is very limited, and there is still no large amount of research on the polymorphic loci of Asians. Therefore, the discovery of the 192-nucleotide polymorphic loci is particularly of far-reaching significance. Toyozumi et al identified the 192nd nucleotide (FⅨ192) in intron 1 as a dinucleotide polymorphic locus, which exists in normal Japanese. 22 In addition, there is no much data about the 192nd locus in the Chinese population. This study included 31 cases, including 4 cases of mutations at this site, namely case 1, case 18, case 19 and case 26. The discovery of these cases not only further verifies the polymorphic locus at this site, but also improves the database of the 192nd nucleotide (FⅨ192) as a dinucleotide polymorphic locus in the Chinese population. From the statistical analysis of sequencing results, we found that all the exons and their flanking sequences of the two HB patients (case 18 and case 19) only had this unique mutation point, which is the polymorphism site. There were no other base changes at other positions, but the patients themselves were diagnosed with HB. We have reason to suspect that the disease-causing mutations may occur in the intron region and non-translated sequences outside the sequenced range. Therefore, for these two cases, we need to do further genetic testing to determine the location of the disease-causing mutations to explain the cause of the disease. The detection methods we can adopt can be to design and synthesize primers covering seven intron fragments and non-translated sequences, optimize the PCR system and conditions, detect them using PCR and direct DNA sequencing methods, or apply other technical means for further testing of these two cases.
According to the sequencing results, by comparing with normal gene sequences through BLAST and checking the latest hemophilia B mutation database, 13 of the 34 mutations detected in this study are newly discovered mutations, which are reported for the first time in the world. This study classifies and analyzes the cases based on the type of mutations. Firstly, in terms of point mutations, we found that four mutations, such as cases 3, 5, 6, and 25, are missense point mutations occurred in exon regions. The amino acid changes are L-Q (leucine-glutamine), Y-D (tyrosine-aspartate), S-P (serine-proline), and V-G (valine-glycine). The amino acid changes in the coding region leads to different degrees of structural and functional changes in the synthesized protein, thereby affecting the activity of coagulation factor proteins in the blood and causing different degrees of coagulation disorders. Clinically, these mutations manifest as B hemophilia with different degrees of bleeding. 23 Secondly, in the deletion type mutations, except for -T as a previously reported mutation point that causes a shift in the reading frame, 24 the other three are newly discovered deletion mutation types. Among them, -GCTGAAACCA deletion (case 14) and -GCTAAACA deletion (cases 16 and 26) are deletion mutation types that cause changes in the reading frame of the coding sequence. The mutation mechanism is that amino acid changes cause protein structure and function changes. 25 Repeated deletion mutations can verify each other's mutation types.
At present, the specific functional mechanism of the shear position in the world is not very clear, and the relevant databases and materials are not complete. As we know, the intron/exon shear position region sequence is an important region bridging between exons. Whether the shear position is normal or not will affect the normal transcription of mRNA, and then determine the normal synthesis of FⅨ protein and ensure its activity in the coagulation system.26,27 Ketterling and others have conducted in-depth research and analysis on this, and their research and analysis show that among 470 mutations, there are 42 mutations that will destroy the normal shear site, with a frequency of 9%. 28 Such situations are similar to the mutation frequency in the coding region, and the consequences of shear site mutations are mostly severe hemophilia. According to the shear site chart, the deletion of -ATTT in case 30 can be judged as a shear site deletion mutation. In addition, T6320A, C6828 T, G6640A, A29753G, G30321 T and T17517G are other 6 mutations that occur in intron regions. Similarly, according to the shear site location, case 4's T6320A occurs at the shear site and can be determined as a pathogenic mutation. It can be seen that these two mutations at the shear site in this study are important findings of HB gene mutations at the shear site location. The other 5 mutations at intron sites are not at the shear site. Whether they are new polymorphisms or pathogenic mutations needs to be further determined using polymorphism site research methods and other genetic diagnosis methods.
In this study, we found that case 15 was a triple mutation patient. The occurrence of each mutation alone can lead to the mechanism of coagulation disorder and lead to the clinical manifestations of bleeding. It can be inferred that the occurrence of triple mutations can lead to more severe coagulation mechanism disorder and more obvious bleeding manifestations, which can be reflected from the lower FIX activity of the venous blood test of this patient. The triple mutation in case 15, −6. G > C of EXON1.35, has been shown to be a pathogenic mutation, 6640.G > A at Ivs2.151 locations, and 29753.A > G at Ivs6.9187 locations, may be polymorphic loci.
In addition to the patients who were detected with gene mutations, there were also some cases that did not get molecular diagnosis. These cases were diagnosed as B hemophilia according to their coagulation activity and clinical manifestations. However, the pathogenic mutations were not found by gene testing, or the mutations were not determined to be pathogenic mutations. Therefore, further experiments were needed to confirm the diagnosis. The gene direct sequencing method used in this study still has some technical limitations, so it needs to be supplemented and improved. The primers in this study covered 8 exons and their flanking sequences, but did not include 7 introns and non-translated sequences at both ends. Therefore, primers were designed to amplify these sequences, and then DNA sequencing was used to detect gene mutations to achieve mutation detection covering the whole gene sequence. As the intron fragment gene sequence is too large, primer design is a challenge. Next, we will conduct experimental research on primer design to achieve the goal of amplifying the target fragment as soon as possible, and then sequencing to find the specific pathogenic mutations.
In the background, the methods of detecting hemophilia have been listed, but various methods have limitations. Some of the diagnosis methods are too complicated and time-consuming, and they only stay in laboratory basic research operations and cannot be applied to clinical molecular diagnosis. With the continuous improvement of living standards and the continuous progress of technology, there is an urgent need to establish a HB diagnosis screening system with increasing understanding of HB. Nowadays, in clinical practical work, multiple polymorphic linkage analysis methods often need to be used in combination to diagnose patients with hemophilia, carriers, and prenatal diagnosis. Indirect linkage analysis methods are simpler, faster, and easier to use without the need to clarify the nature of mutations, but the prerequisite is that the tested family must have a positive family history, at least one proband, and female carriers must be heterozygous at the tested locus. If the polymorphic loci cannot provide information or there is no clear family history, direct gene diagnosis or direct gene sequencing methods still need to be used. Therefore, this study chose direct gene sequencing method for FⅨ gene mutation research, which is convenient, time-saving, and can directly locate mutations and analyze base changes.
In summary, the results of the study of FⅨ gene mutations in 31 patients with HB have important guiding significance for carrier screening and prenatal genetic diagnosis. The development of the times cannot be separated from the improvement of the population quality. Genetic diagnosis plays an important role in reducing the birth rate of HB children and improving the population quality. Clinically, genetic diagnosis and prenatal diagnosis of HB patients and carriers can be carried out by direct gene sequencing combined with linkage analysis. The analysis of gene mutations is the basic basis for genetic diagnosis. This study has made an important addition to the mutation spectrum of the world HB mutation database, and also verified some of the research data on FⅨ gene mutations in HB patients. At the same time, the study of HB gene mutations in the Chinese population has supplemented the research on FⅨ gene mutations in human racial differences, and also provided statistical data for domestic research on HB. With the development of molecular biology, gene therapy has brought hope for radical treatment of HB, and has achieved certain results in clinical applications, but still needs to overcome many gene level difficulties. The analysis of FⅨ gene mutations not only help to further understand the importance of certain amino acid residues in FⅨ gene for FⅨ coagulation activity, but also clarifies its pathogenesis, and provides scientific evidence for future gene therapy.
Conclusion
CpG dinucleotide regions are mutation hotspots.
The 192nd nucleotide of intron 1 (FⅨ192) is a dinucleotide polymorphism locus in the Chinese population.
Point mutations, deletions, and insertions that occur in the exon region and cause amino acid changes or premature termination can be determined as pathogenic mutations. Mutations at splicing sites can also be determined as pathogenic mutations.
For cases with no other gene mutations except for polymorphism loci, sequencing of the exon sequences is required to identify the pathogenic mutation locus. Gene mutations that occur in introns need further identification as pathogenic mutations or polymorphism loci, and sequencing of the exon sequences is also required.
Footnotes
Abbreviations
Acknowledgements
We sincerely thank the expert assistance of the Central Laboratory at the Affiliated Hospital of Putian University.
Author Contributions
Danjuan Liu: conception, design, analysis, and interpretation of the data; writing of the manuscript.
Junting Weng: conception, design, analysis, and interpretation of the data; writing of the manuscript.
Rongjie Guo: analysis, and interpretation of the data; writing of the manuscript.
Bingbing Shi: analysis, and interpretation of the data; writing of the manuscript.
Min Chen: design, analysis, and interpretation of the data; writing of the manuscript.
Zhifang Fu: design, analysis, and interpretation of the data; writing of the manuscript.
Data Availability
The data that support the findings of this study are available from the cooresponding author upon reasonable request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by the Fujian Provincial Health Technology Project (grant number 2023CXA055) and the Natural Science Foundation of Fujian Province of China (grant number. 2020J011253).
Ethics Approval and Consent to Participate
The animal experiments were conducted in accordance with the ARRIVE guidelines and the National Research Council's Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee of the Affiliated Hospital of Putian University, China, approved the experiments (Permit Number: 202013).