
Abstract
Establishing a national screening program for hemophilia patients is highly encouraged by the World Health Organization and the World Federation of Hemophilia. Hence, this study aimed to analyze the variant spectrum of F8 and F9 genes in Arab hemophilia patients. Molecular genetic and sequencing studies were performed on a cohort of 135 Saudi hemophilia patients. Out of all screened hemophilia patients (97 hemophilia A and 39 hemophilia B), 15 (11.1%) were positive for inversion 22 and 4 (3%) for inversion 1. Out of a total of 32 (23.7%) substitution/deletion mutations, 2 novel variants were identified: a novel splice acceptor site missense mutation (c.5816-2A > G) causing a pathogenic variant of the F8 gene and another splicing site point mutation in intron/exon 23 (g.164496G > A). The frequent F8 variants were (c.409A > C, p.T137P) in exon 4, (c.760A > G) in exon 6, and (c.1835G > C, p.R612P) in exon 12, while the frequent F9 variants were (c.580A > G) in exon 6 and (c.880C > T) in exon 8. These study data will enrich the spectrum of the genetic databases in the Arab population that could be applied in the future for national genetic counseling.
Introduction
Hemophilia A (HA) and hemophilia B (HB) (OMIM: 306700 and 306900, respectively) are X-linked recessive bleeding disorders that are caused by the inheritance of genetic variants affecting F8 and F9 genes, those encoding coagulation factors VIII and IX with a consequent deficiency or dysfunction for its relevant factor. 1 The F8 gene sited at the end of the long arm of the X chromosome, at Xq28, is extremely large and structurally complex (186 932 bp; nearly 180-kb length containing 26 exons), while the F9 gene placed more toward the centromere, at Xq27.6, is considerably smaller and structurally simpler (nearly 34-kb length containing only 8 exons). 2
HA is more prevalent than HB worldwide. 3 In 2021, a meta-analysis study suggested that per 100 000 world male population, 17.1 were affected with HA compared to 1.1 affected with HB. 4 Although the clinical manifestations of both HA and HB are indistinguishable, the disease severity and the frequency of bleeding into deep tissues correlate with residual factor activity (Figure 1). The severity of hemophilia is classified depending on the plasma level of factor activity, where the severe form is defined as a factor level less than 0.01 IU/mL (<1% of normal), moderate form as a factor level from 0.01 to 0.05 IU/mL (1%-5% of normal), and mild form with a factor level from 0.05 to 0.3 IU/mL (5%-30% of normal). 1 Severe cases of factor deficiency frequently develop spontaneous bleeding episodes that might be life-threatening with the development of intracranial hemorrhage.

Mutation profile of Arab Saudi HA and HB patients.
For understanding genotype/phenotype variations of hemophilia disorder, an enormous number of genetic abnormalities of F8 and F9 genes including deletions, point mutations, rearrangements/inversions, and insertions have been documented in various international genetic databases relevant to Coagulation Factor Variant Databases supported by the Human Gene Mutation Database (HGMD) 5 and the European Association for Haemophilia and Allied Disorders (EAHAD). 6 However, the relative frequency of these mutations varies between HA and HB. For instance, missense variants are more prevalent mutations in patients with severe HB in contrast to those with severe HA, 5 whereas inversion/gene rearrangement, particularly of intron 22, is the most common defect, being accounted for approximately 50% of the reported molecular abnormalities.3,7
The molecular genetic basis of hemophilia disorder is extremely diverse, being influenced by various modulating factors likely involving genetic and epigenetic variation outside the F8 and F9 genes with subsequent phenotypic variation. 8 The type of genetic mutation in the genes of F8 and F9 is associated with the residual factor activity in plasma and accordingly with the clinically relevant hemophilia phenotypes, where larger gene defects/deletions are commonly allied with a more severe clinical phenotype. 3 However, hemophilia has well-known phenotype heterogeneity that can be observed between patients with the same factor levels or even between patients with the same genetic defect. 9 Moreover, inhibitor antibody development risk in hemophilia-treated patients, particularly with prolonged factor exposure, is influenced by genetic variation, where the risk develops more in severe form with gross genetic defects (large deletion and nonsense mutations) than those with mild and moderate disease.8,10,11 Nevertheless, the incidence of inhibitor risk is higher in HA than in HB with similar severity (25%-30% vs 3%-5%, respectively).
For enriching the hemophilia genotypic/phenotypic database with the nature of variants present in the Arab population that could explain differences in disease phenotypes, this study aimed to analyze the variant spectrum of F8 and F9 genes of hemophilia patients who are registered and regularly followed in 3 different Saudi centers caring for hemophilia as part of a screening program that could be valuable in the future for inherited disease counseling.
Materials and Methods
Patients and Sample Collection
A total of 135 patients with hemophilia (96 HA patients and 39 HB patients) enrolled in this cohort study are registered and regularly followed in different hemophilia programs at the Hematology Clinic in King Faisal Specialist Hospital and Research Center (KFSH & RC) in collaboration with Blood Disorders Center and Maternity and Children Hospital in Al-Madinah Al-Munawarah from 2015 to 2020. Peripheral blood samples were collected in EDTA and citrated vacutainers (BD, Franklin Lakes, NJ, USA) from all patients with informed consent and prior approval of the KFSH & RC ethical committee (RAC 2130036). Molecular genetic analysis of F8 and F9 genes was investigated after taking a detailed clinical history and laboratory testing of factor (VIII and FXI) coagulant activity and inhibitors. Clinical data included bleeding history, type of management using recombinant factor concentrate (prophylactic vs on demand), and inhibitor status at the time of testing for molecular genotype.
The patients were categorized according to clinical severity of the disease as normal coagulant factor activity at 0.5 and 2 IU/mL (>40%), mild cases at 0.05 to 0.4 IU/mL (5%-30%), and moderate form at 0.01 to 0.05 IU/mL (1%-5%), while clinically severe form at <0.01 IU/mL (<1%).12,13
Coagulation Assays and Inhibitor Detection
All collected blood samples into sodium citrate solution (3.2%) were centrifuged and tested for partial thromboplastin time (PTT), factor (VIII and XI) coagulant activity, and factor inhibitors using the chromogenic assay. Stago Diagnostica STA® was used for measuring all coagulation tests. The modified Nijmegen–Bethesda method described by Miller et al 14 was used for estimating the level of inhibitors.
Molecular Genetic Analysis and Sequencing
Genomic DNA was isolated from EDTA's entire blood samples on the MagNa Pure system using the MagNA Pure compact nucleic acid isolation kit-I (Roche, Mannheim, Germany) according to the product's guidelines.
For F8 gene analysis, all samples of HA patients (96) were screened for F8 inversion 22 (inv-22) and inversion 1 (inv-1) before sequencing analysis. For inv-1 and inv-22 testing, polymerase chain reaction (PCR) amplification methods reported by Bagnall et al15,16 were performed. All negative samples for either inv-1 or inv-22 were sequenced for F8 exons 1, 3, 4, 6, 11, 12, 14, 22, and 23 with the flanking intronic regions. While for F9 gene analysis, all 39 samples of patients with HB were amplified and sequenced for all exonic regions of the gene (F9 exons 1-8) as described by Al-Allaf et al. 1 For PCR amplification, the Veriti 96-Well Fast Thermal Cycler (Applied Biosystems, Foster City, CA, USA) using HotStarTaq DNA Polymerase (Qiagen, Hilden, Germany) was used. For designing the sequence of the primers, the Primer3web-based server was utilized (http://frodo.wi.mit.edu/primer3/). For sequencing, the ABI Prism 3730 Genetic Analyzer (Applied Biosystems, Inc.) was used for screening PCR products with the BigDye Terminator v3.1 Cycle Sequencing Kit. For sequence analysis, SeqMan 6.1 (DNAStar Inc., Madison, WI, USA) was used.
The NCBI BLAST program was utilized for data analysis of the sequenced F8 and F9 chromatograms, where the obtained results were compared with the gene reference sequence (GenBank accession numbers NG_2157 for F8 and NG_2158 for F9). In confirming the mutation novelty, the database of ClinVar, dbSNP, HGMD, and EAHAD-CFDB was referred to. Finally, the Human Genome Variation Society (HGVS) nomenclature was used to determine the amino acid sequence of mutated proteins (http://www.hgvs.org/).
Data Statistical Analysis
The study results were analyzed using IBM SPSS Statistics for Windows 21.0 (IBM Corp., Armonk, NY, USA). The Fisher exact test was selected for comparisons, and significant differences were considered when the obtained P value was less than .05.
Results
With the objective intending to elucidate possible genetic variants of F8 and F9 among Saudi hemophilia patients, we studied 135 patients treated at the participating centers. Out of 135 hemophilia patients including 2 symptomatic carriers, 96 (71.1%) were HA and 39 (28.8%) were HB. Their age ranged from 2 to 62 years old (average 22 years old). The clinical severity of HA ranged between mild (9, 9.4%), moderate (5, 5.2%), and severe (80, 83.3%), while for HB, there were 13 (33.3%) mild, 2 (5.1%) moderate, and 24 (61.5%) severe clinical forms. There were 76 cases (55.9%) who had a chronic joint disability. Factor inhibitors with different titers were detected in 24 (25%) of HA and only 2 (5.1%) of HB. (Table 1). The phenotypic/genotypic profile of all detected F8 and F9 variants in our cohort is described in Table 2.
Demographic and Clinical Data of HA and HB Patients at Presentation.
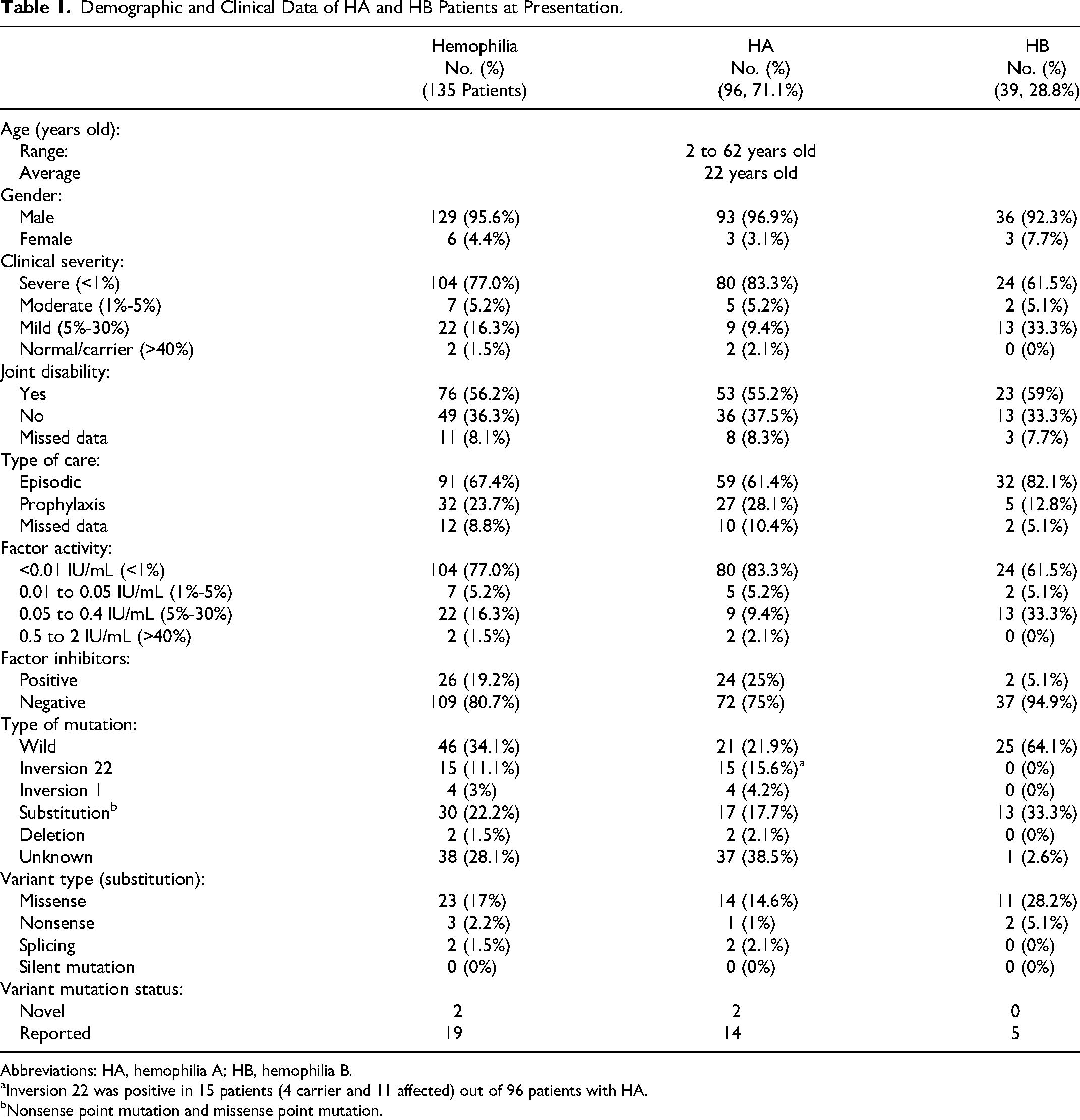
Abbreviations: HA, hemophilia A; HB, hemophilia B.
Inversion 22 was positive in 15 patients (4 carrier and 11 affected) out of 96 patients with HA.
Nonsense point mutation and missense point mutation.
Phenotype/Genotype Profile of 135 Saudi HA and HB Patients.
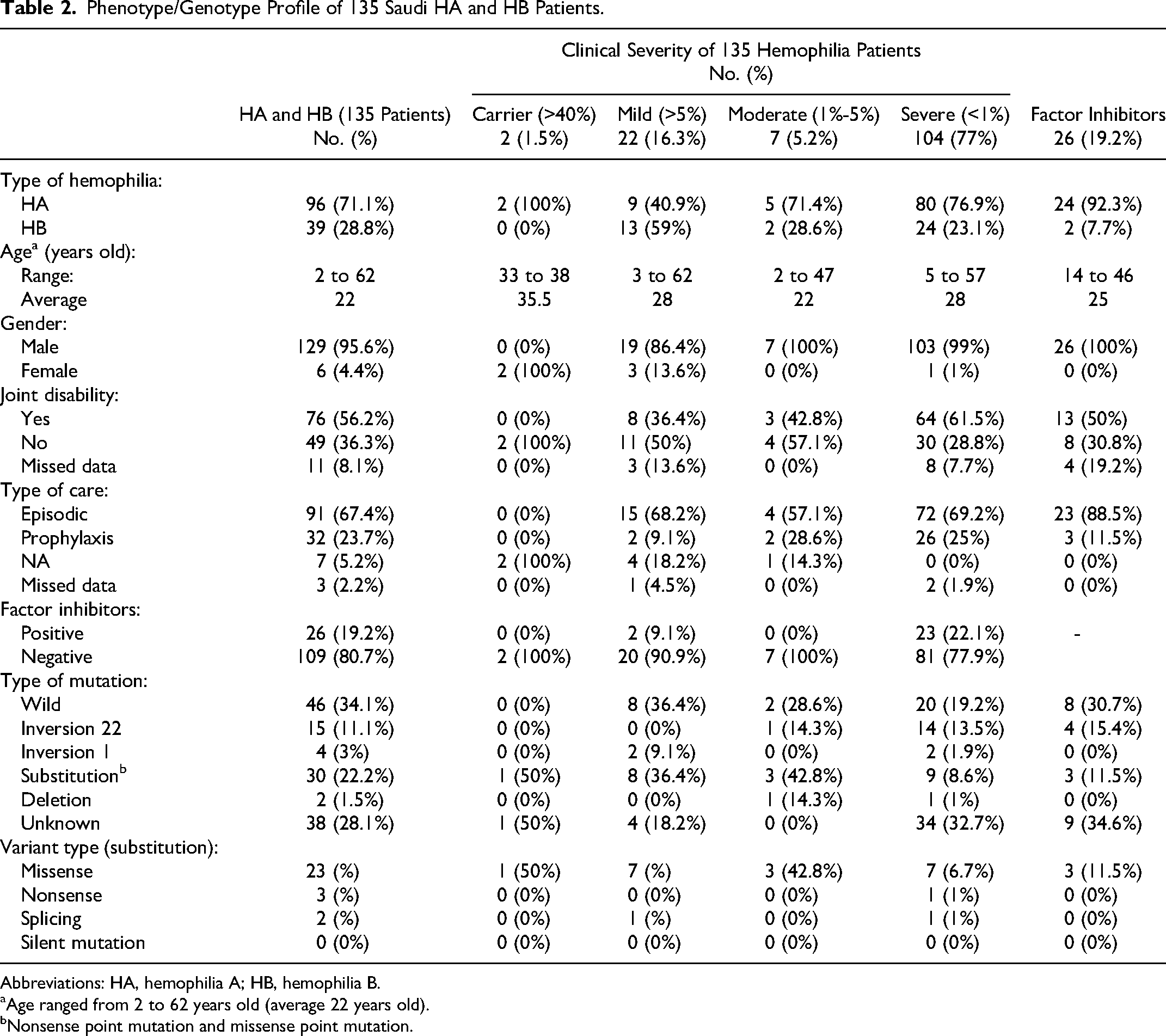
Abbreviations: HA, hemophilia A; HB, hemophilia B.
Age ranged from 2 to 62 years old (average 22 years old).
Nonsense point mutation and missense point mutation.
Out of the whole cohort of 96 HA patients who had been tested for causative variants of the F8 gene, 15 (15.6%) were positive for inv-22, 4 (4.2%) for inv-1, 14 (14.6%) missense, 2 (2.1%) splicing, 1 nonsense, and 2 (2.1%) deletion, and 37 patients (38.5%) had no mutation identified. Regarding F9 gene variants identified in our HB cohort (39), 11 patients (28.2%) had missense mutations, 2 (5.1%) had nonsense mutations, and only 1 case had no mutation identified. Our results were able to characterize 20 already described mutations in the ClinVar database of factor F8 and F9 genes (Table 3). In this cohort study, the frequently detected F8 variants were (c.409A > C, p.T137P) in exon 4, (c.760A > G) in exon 6, and (c.1835G > C, p.R612P) in exon 12, and the frequently detected F9 variants were (c.580A > G) in exon 6 and (c.880C > T) in exon 8 (Supplemental Tables 1 and 2, online version).
Genome Database of FVIII and FXI Variant Types (Substitution/Deletion) Detected Among 135 Arab Hemophilia Patients of This Study.
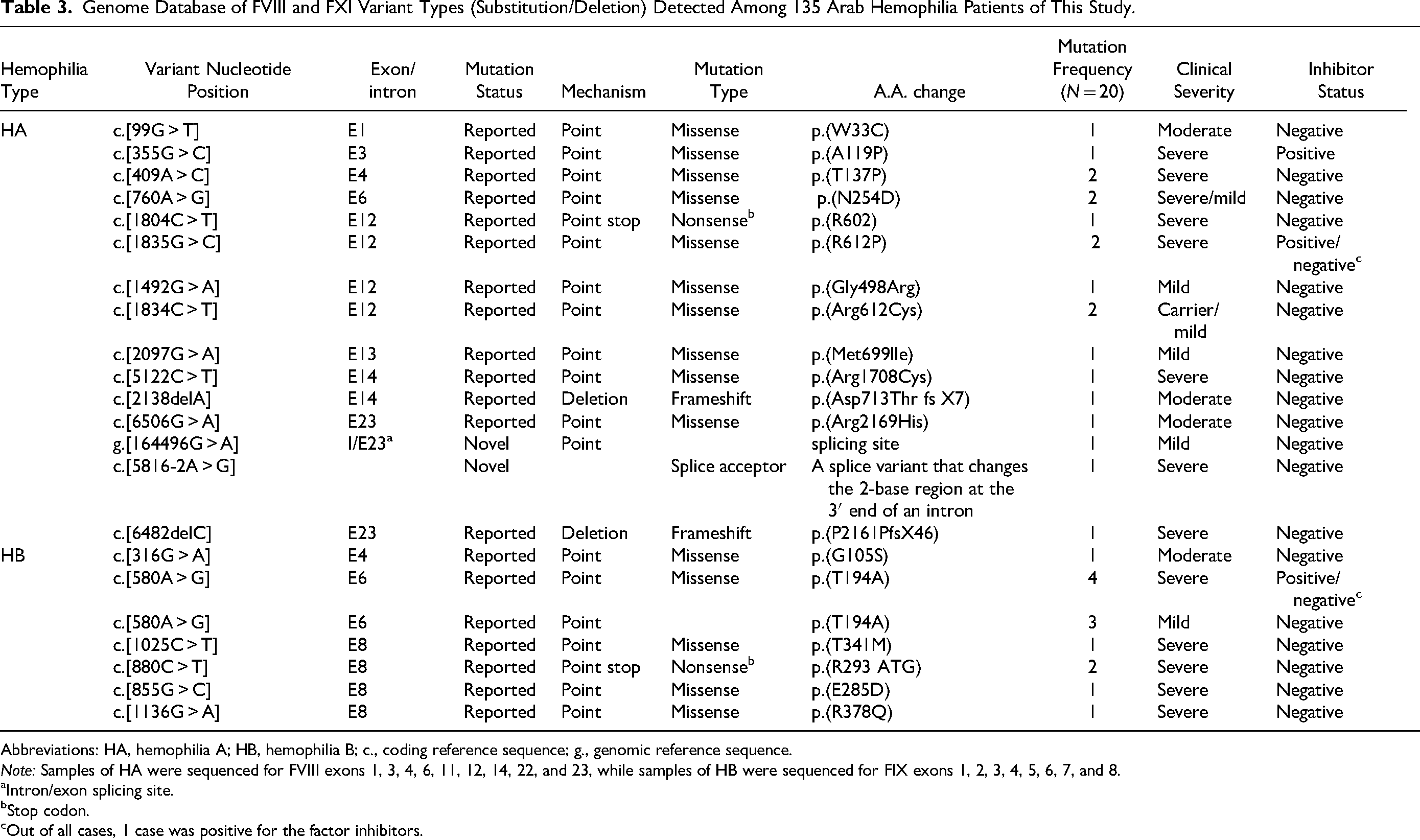
Abbreviations: HA, hemophilia A; HB, hemophilia B; c., coding reference sequence; g., genomic reference sequence.
Note: Samples of HA were sequenced for FVIII exons 1, 3, 4, 6, 11, 12, 14, 22, and 23, while samples of HB were sequenced for FIX exons 1, 2, 3, 4, 5, 6, 7, and 8.
Intron/exon splicing site.
Stop codon.
Out of all cases, 1 case was positive for the factor inhibitors.
Our results were able to characterize 2 novel variants of the F8 gene, to the best of our knowledge, which was not reported previously in the referred genetic database. Phenotype/genotype correlation analysis of our cohort revealed a case with a clinical form more severe than expected by the type of mutation identified, which is a novel splicing acceptor site missense mutation (c.5816-2A > G) causing a pathogenic variant of the F8 gene (severe HA phenotype) with no inhibitor formation detected. In addition, a splicing site point mutation in intron/exon 23 (g.164496G > A) was identified, which correlates well with the severity of the phenotype with no inhibitor formation. For a better understanding of the phenotype–genotype relationship of our cohort, the patients’ detailed phenotype with the novel mutations in F8 was fully described (Table 4).
Clinical Characterization of FVIII Novel Mutations Identified in This Study.

Abbreviations: MS, missense; Fs, frameshift; NA, non-applicable.
Plasma-derived clotting factor concentrate.
Discussion
Despite the vast genotypic records on F8 and F9 variant databases, reports of novel ones continue to enrich the database for a better understanding of the molecular pathogenesis of hemophilia that might elucidate the differences in disease phenotypes and confirm the genotype–phenotype correlation within nations with a related ethnic background. 9 In this study, with the objective intending to analyze the variant spectrum of F8 and F9 genes for boosting the genotypic database with the nature of mutations existing in the Arab population, we studied 135 hemophilia patients referred to KFSH & RC.
According to literature reports on mutation screening of the F8 gene, nearly 30% of detected F8 gene variants are usually novel for the high content of the hyper-mutable GC nucleotides in the region encoding the F8 gene.
17
For this reason, despite the numerous studies published describing different variants in Middle Eastern hemophilia,1,17–21 the data available for the frequent variants causing hemophilia in the nations of this region are still insufficient.
18
In this study, we report 2 novel F8 variants, to the best of our knowledge, which were not published in any up-to-date literature search using the PubMed/Medline database
In line with literature reports, the present study confirms the well-known frequent genetic defect of HA and HB, where the null allele (large deletion, insertion/deletion mutations, nonsense, and gene rearrangements principally inversion of intron 22) was responsible for the most frequent genetic abnormalities in severe HA (10 out of 26 severe forms, 38.4%), whereas missense mutations have been found in the majority of patients with moderate (3 out of 5, 60%) and mild forms of the disease (5 out of 7, 71.4%) and comprise the most frequent molecular defect in HB (>50% of all reported mutations) (Supplemental Tables 1 and 2, online version).7,17,19,20,22,23
Our study also in agreement with previously published data3,24 found that the risk of developing inhibitors was much less frequent in HB patients compared to HA (5.1% vs 25%, respectively), and the risk was allied mostly in severely affected patients except for 2 mild HA cases (Supplemental Tables 1 and 2, online version), thus supporting the assumption that the higher prevalence of the less severe missense mutations in HB compared to HA provides a potential biological reason for explaining the milder disease phenotype regarding bleeding severity and inhibitor risk in HB versus HA. 3 It is well known that the risk for inhibitor development to factor replacement therapy is related to several factors including the type and location of genetic defect, where variants with gross defects, such as gross deletions, inversions breaking, and frameshifts, had the highest risk for inhibitor formation. 25 However, it has been reported that inhibitor occurrence is associated with some missense mutations in the mild form of HA, unlike HB. 3 Although it is renowned that the pathogenesis of inhibitor formation is influenced by genetic predisposing aspects such as ethnicity, type of mutation, human leukocyte antigens (HLA) genotype, severity of hemophilia, and family history of inhibitors, in addition to non-genetic elements such as multiple product switches and onset and duration of replacement therapy, 23 the type of genetic defect is 1 of the most crucial risk factors for inhibitor development.
Out of 135 hemophilia patients included in this cohort, the spectrum of non-inversion F8 mutations included 19 different alterations, namely 14 missense, 1 nonsense, 2 splice site mutations, and 2 insertions/deletions,9,11 while the spectrum of F9 gene mutations comprised 11 (28.2%) missense mutations and 2 (5.1%) nonsense ones. For those 38 cases with unrecognized mutations, their mutations may occur in regions outside the studied genes. 8 To date, out of 3052 unique reported mutations of F8 gene variants in the EAHAD v3.1 database, point substitution was the predominant mutation type (66.2%), followed by deletions, fragment duplication, others, and insertion (23.4%, 6%, 2.8%, and 1.6%, respectively). 25
Furthermore, phenotypic variation in this cohort has been observed between patients with the same molecular genetic defect, confirming the well-known fact that disease severity may be induced by the same mutation.7,9 This finding can be elucidated, where the 2 genetic defects including inv-22 and the missense mutation (c.760A > G) in exon 6 were identified in association with severe and moderate HA disease severities. Additionally, an unexpectedly severe phenotype was noted in a HA case who carries a splice acceptor site missense mutation (c.5816-2A > G) causing a pathogenic variant of the F8 gene. Nevertheless, the type of molecular defect is considered a strong predictor of the disease's clinical phenotype. 9
Regarding the non-inversion frequent variants in our cohort, (c.409A > C, p.T137P) in exon 4, (c.760A > G) in exon 6, and (c.1835G > C, p.R612P) in exon 12 were the frequently identified F8 variants, whereas (c.580A > G) in exon 6 and (c.880C > T) in exon 8 were the frequently recognized F9 variants. The frameshift mutation in exon 23 (c.6482delC, p.P2161Pfs*46) caused a severe form of HA, as reported by Al-Allaf et al. 17
The present study reports 2 novel variants in the F8 gene with different clinical severities, where a splicing acceptor site missense mutation causes a pathogenic variant of the F8 gene in addition to a missense mutation in the splice site in intron/exon 23 mutations (g.164496G > A) correlated well with the severity of the disease phenotype and associated with a mild form of HA. Unlike the former novel mutations described, the novel splicing acceptor site missense mutation (c.5816-2A > G) was unexpectedly causing a pathogenic variant of the F8 gene. This splice variant changes the 2-base region at the 3′ end of an intron causing a severe form of HA with no inhibitor formation detected. In general, point mutations involve nonsense mutations resulting in an early stop codon and premature termination of translation, missense mutations generating amino acid exchange, splicing site defects, and regulation region defects causing alteration of gene expression levels. 7
Incomplete screening for large parts of the F8 gene, where no variant was identified, and the retrospective nature of the data are the limitations of our study. However, these study data will enrich the spectrum of the genetic databases for a better understanding of hemophilia phenotype/genotype patterns within the Saudi population that could be applied in the future for genetic counseling and diagnosis.
Conclusion
In elucidating the phenotypic/genotypic profile of 135 Saudi hemophilia patients, the frequent variants of the F8 gene were (c.409A > C, p.T137P) in exon 4, (c.760A > G) in exon 6, and (c.1835G > C, p.R612P) in exon 12, while the frequent F9 variants were (c.580A > G) in exon 6 and (c.880C > T) in exon 8. Furthermore, we report a novel splicing acceptor site missense mutation (c.5816-2A > G) causing a pathogenic variant of the F8 gene and another splicing site point mutation in intron/exon 23 mutations (g.164496G > A). These study data will enrich the spectrum of the genetic databases in the Arab population that could be applied in the future for national genetic counseling.
Supplemental Material
sj-docx-1-cat-10.1177_10760296231182410 - Supplemental material for Genotype Hemophilia Screening Program Identified 2 Novel Variants Including a Novel Variant (c.5816-2A > G) Causing a Pathogenic Variant of the Factor 8 Gene
Supplemental material, sj-docx-1-cat-10.1177_10760296231182410 for Genotype Hemophilia Screening Program Identified 2 Novel Variants Including a Novel Variant (c.5816-2A > G) Causing a Pathogenic Variant of the Factor 8 Gene by Tarek Owaidah, Salwa Bakr, Nouf Al-Numair, Hala AbaAlkhail, Hazzaa Alzahrani, Mahasen Saleh, Haitham Khogeer, Ahmed Tarawah, Hadeel Akkad and Faisal Al-Allaf in Clinical and Applied Thrombosis/Hemostasis
Footnotes
Author Contributions
Tarek Owaidah, Hala AbaAlkhail, Mahasen Saleh, Ahmed Tarawah, and Hazzaa Alzahrani contributed to patient management and provided clinical data. Nouf Al-Numair, Hadeel Akkad, Haitham Khogeer, and Faisal Al-Allaf contributed to molecular testing, as well as data interpretation and reporting of genetic mutations. Salwa Bakr contributed to data analysis and manuscript writing. All authors contributed to the critical revision of the manuscript and approved its final version.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.